




A unique strategy for overcoming the diffraction barrier employs photoswitchable fluorescent probes to resolve spatial differences in dense populations of molecules with superresolution. This approach relies on the stochastic activation of fluorescence to intermittently photoswitch individual photoactivatable molecules to a bright state, which are then imaged and photobleached. Thus, very closely spaced molecules that reside in the same diffraction-limited volume are temporally separated. Merging all of the single-molecule positions obtained by repeated cycles of photoactivation followed by imaging and bleaching produces the final superresolution image. Techniques based on this strategy are often referred to as probe-based superresolution, and were independently developed by three groups in 2006 and given the names photoactivated localization microscopy (PALM), fluorescence photoactivation localization microscopy (FPALM), and stochastic optical reconstruction microscopy (STORM). All three methods are based on the same principles, but were originally published using different photoswitchable probes.
The tutorial initializes with a number of dark green spheres (representing the dark state of individual fluorophores) appearing under a white curve along the Specimen line. In order to operate the tutorial, use the mouse cursor to move the Scan Progress slider from right to left. As the slider is moved to the right, individual spheres become excited (turn red) and are transiently recorded in the CCD Image Plane before being permanently assigned as a location on the Image. Clicking on the Autoscan button will transition through the tutorial automatically and the Reset button can be used to re-initialize the tutorial.
The principle surrounding photoactivated localization microscopy and related techniques rests on a combination of imaging single fluorophores (single-molecule imaging) along with the controlled activation and sampling of sparse subsets of these labels in time. Single-molecule imaging was initially demonstrated in 1989, first at cryogenic temperatures and later at room temperature using near-field scanning optical microscopy. Since that time, the methodology has evolved to being a standard widefield microscopy technique. The prior knowledge that the diffraction-limited image of a molecule originates from a single source enables the estimation of the location (center) of that molecule with a precision well beyond that of the diffraction limit. This high level of precision is scalable with the inverse square of the number of detected photons, as will be described below.
In general terms, a single fluorescent molecule forms a diffraction-limited image having lateral and axial dimensions defined by the excitation wavelength, refractive index of the imaging medium, and the angular aperture of the microscope objective:
Resolutionx,y = λ / 2[η • sin(α)](1) Resolutionz = 2λ / [η • sin(α)]2(2)
where λ is the wavelength of light (excitation in fluorescence) and the combined term η • sin(α) is known as the objective numerical aperture (NA). Objectives commonly used in microscopy have a numerical aperture that is less than 1.5 (although new high-performance objectives closely approach this limit), restricting the term α in Equations (1) and (2) to less than 70 degrees . Therefore, the theoretical resolution limit at the shortest practical excitation wavelength (approximately 400 nanometers when using an objective having a numerical aperture of 1.40) is around 150 nanometers in the lateral dimension and approaching 400 nanometers in the axial dimension. In practical terms for imaging of enhanced green fluorescent protein (EGFP) in living cells, these values are approximately 200 and 500 nanometers, respectively. Thus, structures that lie closer than 200 to 250 nanometers cannot be resolved in the lateral plane using either a widefield or confocal fluorescence microscope.
When examining the lateral image plane in a viewfield of single molecule emitters, the central portion of each diffraction-limited spot corresponds to the probable position of a molecule and can be localized with high nanometric precision by gathering a sufficient number of photons. Methods for determining the center of localization are based on a statistical curve-fitting of the measured photon distribution (in effect, the image spot, but also referred to as the point-spread function) to a Gaussian function (see Figure 1). The ultimate goal in this exercise is to determine the center or mean value of the photon distribution (μ = x0,y0), and its uncertainty, the standard error of the mean, σ, according to the equation:
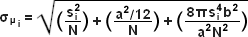 (3)
(3)
where N is the number of photons gathered, a is the pixel size of the imaging CCD detector, b is the standard deviation of the background (which includes background fluorescence emission combined with detector noise), and si is the standard deviation or width of the distribution (in direction i). The index i refers to either the x or y direction. The first term under the square root on the right-hand side of Equation (3) refers to the photon noise, whereas the second term encompasses the effect of finite pixel size of the detector. The last term takes into account the effect of background noise. By applying these simple mathematical techniques, a localization accuracy of approximately 10 nanometers can be achieved with a photon distribution of around 1000 photons when the background noise is negligible. Extension of this insight has resulted in a host of clever experiments studying isolated structures that are separated by less than the diffraction limit, such as labeled molecular motors and nanoscale DNA molecular "rulers". A major technological development in supporting probe-based superresolution is the widespread availability and ease of use of EMCCD camera systems (see Figure 2), which have single-photon sensitivity.
As described by Equation (3), the key to high-precision results for molecular localization in single-molecule superresolution imaging is to minimize background noise and maximize photon output from the fluorescent probe, a task easier described than accomplished. In the best case scenario, if 10,000 photons can be collected in the absence of background before the fluorophore bleaches or is switched off, the center of localization can be determined with an accuracy of approximately 1 to 2 nanometers. In contrast, if only 400 photons are gathered, the localization accuracy drops to around 20 nanometers or worse. Background in superresolution specimens arises from natural or transfection reagent-induced autofluorescence, as well as from residual fluorescence of surrounding probes that have entered the dark state. Thus, for probe-based single-molecule superresolution imaging techniques such as PALM, the fluorescent molecules should display a high contrast ratio (or dynamic range), which is defined as the ratio of fluorescence before and after photoactivation. Variability in contrast ratios in fluorescent proteins and synthetic fluorophores is due to differences in their spontaneous photoconversion in the absence of controlled activation.
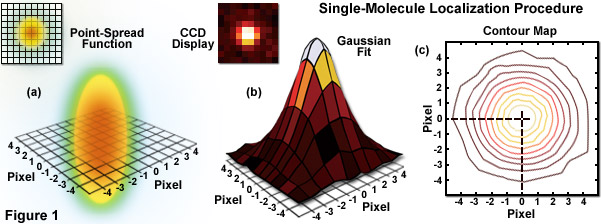
Illustrated in Figure 1 are the steps involved in localizing single molecules with high precision by fitting the point-spread function to a Gaussian function. In Figure 1(a), the point-spread function of a widefield fluorescence microscope is superimposed on a wireframe representation of the pixel array from a digital camera in both two (upper left) and three-dimensional diagrams. The pixelated point-spread function of a single fluorophore as imaged with an EMCCD is shown in the upper left of Figure 1(b), and modeled by a three-dimensional Gaussian function, with the intensity for each pixel color-mapped in the central portion of Figure 1(b). A contour map of the intensities is presented in Figure 1(c). In cases where two contour maps overlap due to emission by fluorophores with a separation distance shorter than the diffraction limit, the centroid for each fluorophore can be individually localized by subtracting the point-spread function of one fluorophore from the other (after it enters a dark state or is photobleached) due to the temporal mapping strategy for generating PALM images.
Most of the structures and organelles observed by fluorescence microscopy in biological systems are very densely populated with fluorophores, with significantly more than one molecule sharing the same diffraction-limited volume. In such cases, single-molecule localization is virtually impossible due to the fact that resulting fluorescence images appear as a highly overlapping distribution of blurred diffraction-limited spots. The mechanism around this predicament, and central to the concept surrounding PALM, is to sequentially localize sparse subsets of molecules. This concept was first demonstrated by Eric Betzig and Harald Hess a decade before PALM was introduced by resolving densely distributed luminescent centers (having distinct spectra) in a quantum well by imaging with a finely binned wavelength. As a result, although there were many centers confined within a spatial resolution volume, they became resolvable when separated along another dimension, wavelength.
The generalization of Betzig and Hess' experiment for microscopy is that if the fluorescent response from an ensemble of molecules can be spread out in some higher dimensional space, then it would be possible to image the sources individually, localize each of their centers, and then accumulate the center coordinates to create a superresolution image. The concept was successfully applied in several investigations prior to the introduction of PALM and STORM. Using laser spectroscopy, the spectra (and localization) of overlapping organic fluorophores was successfully separated, albeit at cryogenic temperatures. Subsequently, the methodology was extended to single-molecule localization through sequential photobleaching or by stochastic recruiting of fluorescent probes to a stationary binding site. Additionally, single-molecule localization has been demonstrated through the analysis of the stochastic blinking of quantum dots.
Probe-based superresolution microscopy became a reality with the discovery of photoactivatable fluorescent labels (both fluorescent proteins and synthetic fluorophores) that could be sequentially switched on in time to produce thousands of sparse subsets. The operating principle of this methodology is to start with the vast majority of fluorescent labels in the inactive state and not contributing to specimen fluorescence. A small fraction (less than 1 percent) is then activated by illumination with near-ultraviolet light (in some cases other spectral regions are useful) to induce a chemical modification in a few molecules that enables them to fluoresce. That sparse subset is then imaged and localized to generate coordinates having nanometer-level precision (see Figure 1). The registered labels are then removed by photobleaching so that a new sparse subset can be transferred into the active state and sampled to gather a new set of molecular coordinates. The process can be repeated many thousands of times up to the point that millions of such molecular coordinates are accumulated within the region being imaged. A composite image rendered from all the coordinate sets produces a single-molecule superresolution image of the fluorescently-labeled structure under investigation. Note that the molecular coordinates are not actually represented by a single point to identify their spatial position, rather a Gaussian intensity distribution corresponding to the positional uncertainty of their location is employed to build the image map.
Contributing Authors
Stephen P. Price and Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.