
Introduction
Although the first report of fluorescence emission in the bioluminescent hydrozoan jellyfish species Aequorea victoria was recorded in the mid-twentieth century and a protein extract was independently demonstrated by two investigators to be responsible for this "green" fluorescence in the 1960s and 1970s, it took another 20 years and a number of significant advances in the technology of molecular and cellular biology to witness the elucidation of the primary amino acid structure. Following shortly thereafter was the astonishing demonstration that the jellyfish green fluorescent protein (GFP; Figure 1) could be readily employed as a useful marker for gene expression in cells evolutionarily far removed from the jellyfish.
During the next several years, a number of "enhanced" genetic variants featuring fluorescence emission spectral profiles spanning the blue, cyan, green, and yellow regions of the visible spectrum were developed by engineering specific mutations into the original GFP nucleotide sequence. One of the most significant advances following the initial cloning and early mutagenesis efforts on the Aequorea victoria green fluorescent protein was the discovery of similar proteins in non-bioluminescent reef corals and sea anemones, which not only provided a large spectrum of new emission colors, but also demonstrated that these protein motifs occur in a wide range of organisms.
Over the past decade, fluorescent proteins have launched a new and unprecedented era in cell biology by enabling investigators to apply routine molecular cloning methods, fusing these optical probes to a wide variety of protein and enzyme targets, in order to monitor cellular processes in living systems using fluorescence microscopy and related methodology. The spectrum of applications for fluorescent proteins ranges from reporters of transcriptional regulation and targeted markers for organelles and other subcellular structures to fusion proteins designed to monitor motility and dynamics. These fascinating probes have also opened the door to creating biosensors for numerous intracellular phenomena, including pH and ion concentration fluctuations, protein kinase activity, apoptosis, voltage, and cyclic nucleotide signaling. By applying selected promoters and targeting signals, fluorescent protein biosensors can be introduced into an intact organism and directed to a host of specific tissues, cell types, as well as subcellular compartments to enable an unprecedented focus on monitoring a variety of physiological processes.
When coupled to recent technical advances in widefield fluorescence and confocal microscopy, including ultra-fast low light level digital CCD cameras, spinning disk, structured illumination, and swept-field instruments, as well as multitracking laser scanning systems with acousto-optic tunable filter (AOTF) control, the green fluorescent protein and its color-shifted genetic derivatives have demonstrated invaluable service in many thousands of live-cell imaging experiments. Compared to many traditional synthetic fluorophores, which are often toxic or photoreactive, the use of fluorescent proteins is minimally invasive for living cells, enabling visualization and recording of time-lapse image sequences for extended periods of time. Furthermore, continued advances in genetically fine-tuning the properties of fluorescent protein variants have led to increased brightness levels, improved photostability, and significantly better expression in mammalian cells. These functional enhancements have stimulated a wide variety of investigations into protein dynamics and function using fluorescent protein chimeras imaged at low light intensities for many hours to extract valuable information about changes in the steady-state distribution.
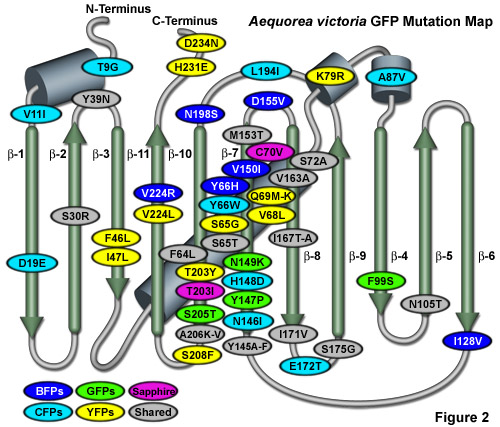
Illustrated in Figure 2 is an Aequorea victoria GFP mutation map showing many of the common mutations superimposed on a topological layout of the peptide structure. The beta-sheets are numbered and depicted as thin, green cylinders with an arrow pointing towards the C-terminus, whereas alpha-helices are depicted by blue barrels. Mutations are color-coded to represent the variants to which they apply: BFPs (blue), CFPs (cyan), GFPs (green), YFPs (yellow), and Sapphire (violet). Folding, shared, and monomerizing mutations are indicated with gray ellipses. Note that almost 75 percent of the mutations are located in the central helix and beta-sheet strands 7, 8, and 10. In general, wavelength-specific mutations occur near the central helix containing the chromophore, while folding mutations occur throughout the sequence.
Despite all of the new and exciting advances in fluorescent protein technology, most of the research groups around the world are still using the enhanced version of wild-type GFP (termed EGFP), as well as the original cyan and yellow derivatives, as workhorses for a majority of the published investigations in cell biology. The reluctance of many researchers to transition to newer fluorescent protein variants is fueled by the checkered availability of these proteins, coupled with (the all too often justified) uncertainties about reported claims of improved brightness, photostability, monomeric character, and utility in fusions. In many cases, just finding a source for a new fluorescent protein or fusion vector of potential interest can be a time-consuming and discouraging challenge. The lack of dependable commercial availability often requires researchers to rely on the generosity of the original source laboratory, which in turn, can be bombarded with an avalanche of requests shortly after a new protein, biosensor, or fusion construct has been reported. Barring the implementation of an organized and efficient system for the distribution of fluorescent protein variants to the scientific community, the situation is unlikely to be remedied in the near future.
Those researchers who are ultimately able to obtain new fluorescent protein derivatives can be seriously disappointed with the results when they compare the new protein to their standard GFP derivatives. The fusion constructs are often hampered by aggregation, low brightness levels, and rapid photobleaching, or may not target a particular organelle as successfully as the classical Aequorea GFP variants. It is not unusual for the protein developers to get feedback in the form: this new (choose a color) protein forms aggregates in the cytoplasm, doesn't form the correct (choose a target) structure as well, and is much dimmer than our other GFP constructs--we can't really use it for multicolor imaging as we expected. Do you know of something better? Actually, the situation is not universally this dismal and the new fluorescent protein derivatives, which seem to be reported every several weeks, hold tremendous promise as versatile reporters for advanced imaging technologies in a number of arenas, regardless of the various potential pitfalls.
Green Fluorescent Protein Structure
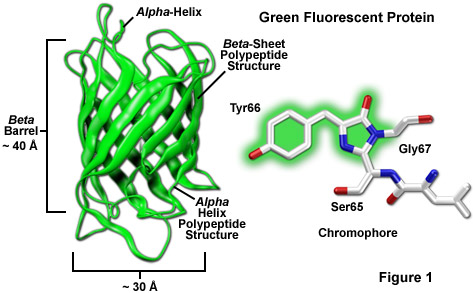
Among the most noteworthy attributes of the original green fluorescent protein derived from the Aequorea victoria jellyfish, as well as the more recently developed palette of color-shifted genetic variants, is that a stable and highly-defined cylindrical polypeptide structure is essential for the development and maintenance of fluorescence in this very remarkable family of proteins. The principle fluorophore (most often termed a chromophore) in GFP is a tripeptide consisting of the residues serine, tyrosine, and glycine at positions 65-67 in the sequence. Although this simple amino acid motif is commonly found throughout nature, it does not generally result in fluorescence. The chromophore forms spontaneously after translation without the requirement for cofactors or external enzyme components (other than molecular oxygen), through a self-catalyzed intramolecular rearrangement of the tripeptide sequence to produce the fluorescent species (as illustrated in Figures 1 and 3 for the naturally occurring wild-type and enhanced GFP variant, respectively).
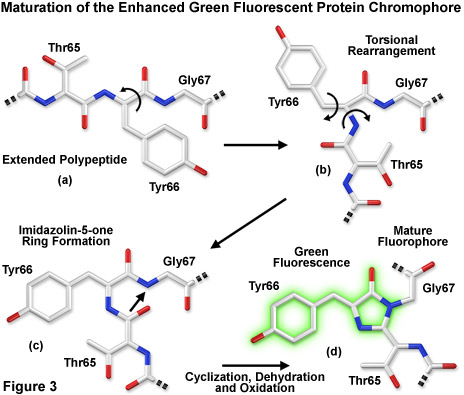
Presented in Figure 3 is a schematic diagram of the chromophore formation in maturing enhanced green fluorescent protein (EGFP), a derivative that substitutes the amino acid threonine for serine at position 65 (S65T). In Figure 3(a), the pre-maturation EGFP fluorophore tripeptide amino acid sequence (Thr65-Tyr66-Gly67) is stretched into a linear configuration so that the threonine residue is positioned in the upper left-hand corner of the diagram. The first step in the maturation sequence is thought to be a series of torsional peptide and side-chain bond adjustments that relocate the carboxyl carbon of the serine amino acid at position 65 (Ser65) in close proximity to the amino nitrogen atom of the glycine residue at position 67 (Gly67; see Figures 3(b) and 3(c)). Nucleophilic attack by this carbon atom on the amide nitrogen of glycine, followed by dehydration, results in the formation of a conjugated imidazolin-5-one heterocyclic ring system (Figure 3(c)). Fluorescence occurs when oxidation of the tyrosine (Tyr66) alpha-beta carbon bond by molecular oxygen extends electron conjugation of the imidazoline ring system to include the tyrosine phenyl ring and its para-oxygen substituent (Figure 3(d)). The result is a highly conjugated π-electron resonance system that largely accounts for the spectroscopic properties of the protein. Extensive mutagenesis studies suggest that the glycine residue at position 67 is critical in formation of the chromophore, and indeed, no fluorescent proteins have yet been discovered that lack this essential element.
The protective beta-barrel (described in more detail below) structure provides a scaffold surrounding the fluorophore to maintain planarity and foster a wide range of interactions with trapped water molecules and amino acid side chains from the polypeptide backbone. Combined with the short portions of alpha-helix and loops at the ends of the barrel (Figure 1), the entire structure serves as a shield against environmental damage to the fluorophore. In this regard, the requirement for molecular oxygen as a fluorophore activation catalyst to form the extended aromatic system in fluorescent proteins is remarkable considering that oxygen must ultimately be excluded from regular interactions with the fluorophore to avoid collisional quenching of fluorescence. The generally low photobleaching rate of fluorescent proteins suggests that the design has evolved with a sacrifice of efficient fluorophore formation as a compromise for long-term stability and higher quantum yields.
Several fundamental physical and chemical aspects of the fluorescent protein chromophore have profound implications for the application of these biomolecules as intracellular probes. A striking aspect of the fluorescent protein chromophores is that although their presence to date has only been detected in sea creatures, they are able to form functional units in a wide variety of organisms ranging from bacteria and plants to mammals. The native Aequorea victoria GFP is composed of a single-domain protein with 238 amino acids and a molecular weight of approximately 27 kiloDaltons. Even though the photophysics of GFP are rather complex, the molecular structure of the protein is quite robust and can accommodate a high degree of modification (executed through the amino acid sequence) without destroying the ability to emit fluorescence.
Fluorescent proteins, or at least chromoproteins bearing the ubiquitous 11-stranded beta-barrel structure capable of producing fluorescent proteins through mutagenesis, have been discovered in organisms ranging from marine invertebrates to crustaceans and probably exist in many other species. In fact, a protein known as nidogen, tucked away in basement membrane of all mammals, has been characterized to have a domain containing a three-dimensional structure remarkably similar to GFP, despite having only 10 percent sequence homology. In nidogen, the amino acid triplet Ile-Gly-Gly (IGG) replaces the chromophore-forming residues Ser-Tyr-Gly (SYG) found in wild-type GFP. In addition, several other residues that are critical for the generation of a functional chromophore in fluorescent proteins have been replaced in nidogen by residues that eliminate the possibility of fluorescence. Nevertheless, the beta-barrel structure appears to have been evolutionarily conserved for a variety of purposes other than fluorescence and nature may surprise us once again with a new source of chromoproteins and fluorescent proteins in species previously not considered.
A wide variety of amino acid substitutions to the original configuration have been successful in fine-tuning the fluorescence of native GFP to provide a broad range of derivative fluorophores that emit colors traversing from the blue to the yellow regions of the visible spectrum (see Figure 2). Site-directed and random mutagenesis investigations have revealed that fluorescence is very dependent on the three-dimensional structure of amino acid residues surrounding the chromophore. Denaturation of the protein, as might be expected, destroys fluorescence, and mutations in residues immediately adjacent to the chromophore can significantly alter the fluorescent properties. Surprisingly, amino acid substitutions in regions of the polypeptide far removed from the chromophore can also affect the spectral characteristics of the protein.
The remarkably well-conserved cylindrical geometry of all the fluorescent proteins discovered thus far appears to be ideally suited to the primary function of protecting the chromophore. Formed in the shape of a cylinder having dimensions of approximately 30 x 40 Angstroms (see Figure 1), the polypeptide backbone is wound into 11 strands of an extensively hydrogen bonded beta-sheet that surround a central alpha-helix containing the chromophore with short helical segments protruding from the ends of the cylinder. Dubbed a beta-can (or beta-barrel) because of the geometrical symmetry, the tight packing of amino acid residues bestows a high level of stability to the protein, which often results in relatively large fluorescent quantum yields for GFP and its derivatives (up to 80 percent).
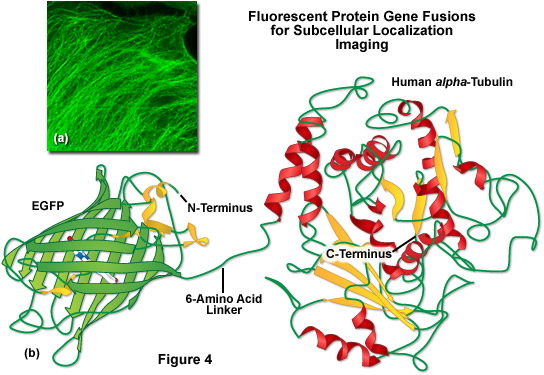
A lack of clefts and gaps for access of small ligands (such as ions and oxygen), combined with the fact that the chromophore is located near the exact center of the protein, partially explains the extraordinary photostability and high quantum yields that are observed. In addition, both the N and C termini are "conveniently" exposed on the surface of the beta-barrel to readily participate in protein fusions that do not affect the structural integrity (Figure 4). This consolidated protein organizational motif also enhances resistance to changes in pH, temperature, fixation with paraformaldehyde, and the disruptive action of many common denaturing agents, such as urea and guanidine hydrochloride. Mutations in the amino acid sequence that affect the fluorescence profile generally also produce negative effects on overall stability, usually resulting in a reduction of quantum yield, increased rates of photobleaching, and enhanced environmental sensitivity.
Illustrated in Figure 4(b) is a drawing of a fusion construct of EGFP with human alpha-tubulin, one of the common subcellular localization probes in widespread use for examining microtubule dynamics. A short peptide linker containing six amino acid residues connects the C-terminus of EGFP to the N-terminus of alpha-tubulin, and the entire unit is transcribed and translated as a single polypeptide with each domain assembling independently. In the ideal fusion, the fluorescent protein is sufficiently well separated from the targeting protein (with a short amino acid linker) so that it does not interfere with normal protein function. In the case of alpha-tubulin, the EGFP chimera localizes correctly to microtubules (Figure 4(a)), but has been shown to hamper normal tubulin dynamics in some investigations. Fusions to alpha-tubulin require an absence of oligomerization for the attached fluorescent protein and thus, are an excellent construct to test for monomeric character.
Fluorescent Protein Spectral Properties
Within the fluorescent protein beta-barrel structure, the cavity containing the chromophore is lined with several charged amino acid residues in the immediate vicinity of the chromophore and contains water molecules that are important in establishing a hydrogen-bonding network surrounding the chromophore. This network of charged amino acid side chains and water molecules, along with van der Waals, π-orbital stacking, and hydrophobic interactions with other amino acids in the area, is critical in establishing the spectroscopic and dynamic photochemical properties of GFP and its derivatives. Mutations that directly alter the covalent structure of the chromophore, or that significantly change the surrounding microenvironment, usually have a profound effect on the spectroscopic properties (absorption and emission profiles) of fluorescent proteins. Many of the mutations that favorably affect spectroscopic and folding properties of GFP derivatives are located in about 20 residues spaced throughout the sequence (see Figure 2), but are usually relatively conservative substitutions. Although a wide variety of mutations result in a total loss of fluorescence, presumably due to chromophore inactivation, most remain uncharacterized even though a careful investigation could potentially assist with the elucidation of residues important in folding and chromophore formation.
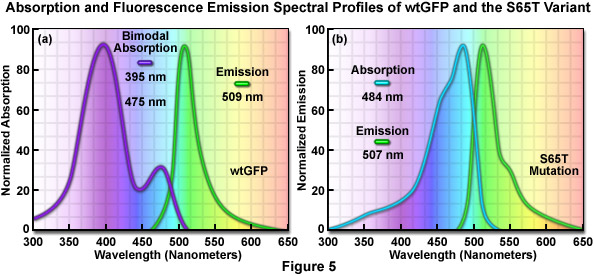
The original wild-type GFP, as isolated and purified from the Aequorea victoria northern Pacific jellyfish, has a principal absorption band peaking at 395 nanometers with a minor band having a maximum at 475 nanometers accompanied by a broad shoulder around 490 nanometers (Figure 5(a)). The extinction coefficient ratio between these ultraviolet and blue absorption spectral peaks is approximately 3:1. Excitation of wild-type GFP with ultraviolet light at 395 nanometers produces fluorescence emission with a maximum at 509 nanometers, while irradiation of the protein with cyan-blue 488-nanometer light results in slightly shorter emission wavelengths with a maximum at 503 nanometers. Because the emission peak position depends on the excitation wavelength, at least two chemically distinct species must be present in wild-type GFP. The origin of the bimodal absorption and dual emission phenomena is attributed to the existence of an equilibrium between two protonation states of the chromophore (neutral and anionic). The peak at 395 nanometers is due to the neutral form of the chromophore and the peak at 475 nanometers occurs as a result of an anionic form, while the 490-nanometer shoulder represents a zwitterionic species. The anionic and neutral species exist in a ground-state equilibrium that can be altered by changes in protein concentration, ionic strength, pH, and temperature.
The similar emission spectra produced by selective excitation of either the neutral (395 nanometers) or anionic species (475 nanometers) of wild-type GFP is credited to the fact that the phenolic oxygen of the chromophore tyrosine at position 66 (Tyr66; Figures 1 and 3) is more acidic in the excited state than in the ground state. Thus, due to the phenomenon known as excited-state proton transfer (ESPT), a common anionic excited state occurs in wild-type GFP and is responsible for the fluorescence emission properties. As light absorption and emission is generated in cycles by periodic excitation, the proton transfer eventually reverses as irradiation is ceased. However, upon radiation with high-intensity ultraviolet light, a percentage of the chromophores remain ionized (photoisomerized) and the 395-nanometer absorption maximum decreases with a concurrent increase in the 475-nanometer peak. Before illumination, wild-type GFP has a ratio of approximately six neutral to each anionic species, but with sufficient ultraviolet illumination, the percentage of anionic molecules can increase several fold. In many biological applications, the coexistence of mixed chromophores having two excitation peaks has many disadvantages. Most notable among these is that the photoisomerization effect significantly diminishes the ability to gather quantitative images.
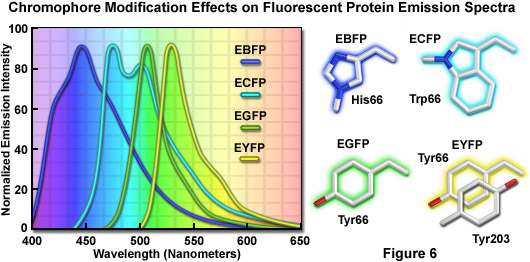
The discovery that substitution in the wild-type GFP chromophore of threonine for serine at position 65 (S65T; known as enhanced green fluorescent protein, EGFP) produced a dramatically improved variant with simple excitation and emission spectra (Figure 5(b)) led to a new class of brighter fluorophores far more suited for biological investigations. In the enhanced GFP mutant, which is twice as bright as wild-type GFP, the absorption spectrum features a single peak at 484 nanometers with an emission maximum at 507 nanometers. Furthermore, these spectral profiles are not altered upon prolonged or repeated illumination. Another advantage of EGFP is that the fluorophore can be readily excited with the popular and common 488-nanometer laser and fluorescence can be monitored with standard FITC filter sets (as described below). A variety of enhanced variants, ranging in emission color from blue to yellow-green, have been produced from derivatives of the original Aequorea victoria protein (see Figure 6). In general, the absorption and emission spectra of these proteins are very broad and featureless, usually spanning a wavelength band ranging from 100 to 200 nanometers. Although these broad spectral profiles allow for great latitude in choosing excitation and emission filters for fluorescence microscopy, they also increase the level of spectral bleed-through when two or more fluorescent proteins are combined in multicolor imaging experiments.
Among the many problems associated with using fluorescent proteins in live-cell imaging is choosing the appropriate color. An important consideration with respect to color selection is the greater desirability of red-shifted fluorophores. It is generally accepted that excitation with violet or blue light is associated with greater cellular phototoxicity than excitation with green, yellow, or longer wavelength light extending through the near infrared (up to approximately 1000 nanometers) but not into the true infrared (where heating due to absorption by water would be problematic for cell viability). Fluorescence excitation and emission hues of fluorescent proteins are confined to a relatively narrow region of the electromagnetic spectrum (essentially the visible wavelengths) due to protein-imposed restrictions on the possible manipulations of the chromophore structure and environment. In contrast, synthetic dyes and nanoparticles with fluorescence emission tuned to wavelengths that cover the visible and near infrared regions of the spectrum are commercially available. This spectral limitation of fluorescent proteins is exacerbated by their relatively broad excitation and emission peaks, which further restrict the number of colors that can be distinguished with bandpass filters on a widefield microscope.
Practically speaking, the bandwidth of the absorption and emission peaks is an important consideration in defining the number of colors that are spectrally distinct, and therefore can be used simultaneously in multicolor imaging scenarios. Roughly speaking, there are currently about 10 different emission colors of fluorescent proteins with short Stoke shifts (defined as the distance in nanometers between the absorption and emission peak wavelengths of a fluorophore) and emission maxima spaced every 20 nanometers between 450-650 nanometers. These colors include: blue (~450 nanometers), cyan (~470 nanometers), teal (~490 nanometers), green (~510 nanometers), yellow (~530 nanometers), yellow-orange (~550 nanometers), orange (~570 nanometers), orange-red (~590 nanometers), red (~610 nanometers), and far-red (>630 nanometers). There are a few additional long Stoke shift fluorescent proteins such as Sapphire and Keima, which should be considered as additional color classes. However, due to the relatively broad excitation and emission peaks shared by all fluorescent proteins, it is only really practical to simultaneously image three (Figure 9) or four distinct colors (such as cyan, yellow, and red or blue, green, orange, and far-red) using a bandpass filter-based microscopy system. However, this tenet doesn't always hold true as the imaging of six distinct colors has been achieved using a single laser line for excitation and spectral unmixing of the emission.
Fluorescent Protein Brightness and Maturation
When designing experiments using fluorescent protein reporters, investigators should ensure that the highest possible signal levels are achieved, especially in cases where the targeted species is rapidly turned over or exhibits low overall population abundance. The apparent brightness of a fluorescent protein, a seemingly qualitative concept, is determined by a number of factors that include the efficiency and rate of maturation, the level of protein expression, the molar extinction coefficient values within the excitation wavelength range, and the quantum yield. In addition, the fluorescence filter combination, spectral distribution of the illumination source, and the digital camera parameters utilized for imaging of fluorescent proteins are of paramount importance in determining whether adequate contrast and signal-to-noise ratios can be achieved.
The intrinsic brightness of a fluorescent protein is determined by the product of the molar extinction coefficient at the peak of the absorption band and the integrated emission quantum yield. As an example, if two proteins have identical quantum yields but one has twice the extinction coefficient at the wavelength of excitation, then the protein with the largest extinction coefficient would have double the brightness of the protein with the smaller extinction coefficient. Likewise, in fluorescent proteins with similar molar extinction coefficients, the one with the highest quantum yield will be the brightest. Quantitative assessment of extinction coefficients and quantum yields is a tedious process that requires a highly purified and correctly folded protein with greater than 95 percent of the molecules having an active fluorescent chromophore. In addition, the total protein concentration must be accurately determined and the measurements of absorption and fluorescence emission performed in a reliable, calibrated spectrophotometer and fluorimeter. Quantum yield assessment requires the comparison of emission spectra between the unknown fluorescent protein and a reference standard having a similar wavelength profile. Investigators should be skeptical of purely qualitative fluorescent protein brightness evaluations (often made by commercial distributors) that lack quantitative information pertaining to the extinction coefficient and quantum yield. It is difficult, if not impossible, to accurately perform brightness comparisons between fluorescent proteins without knowledge of these critical parameters.
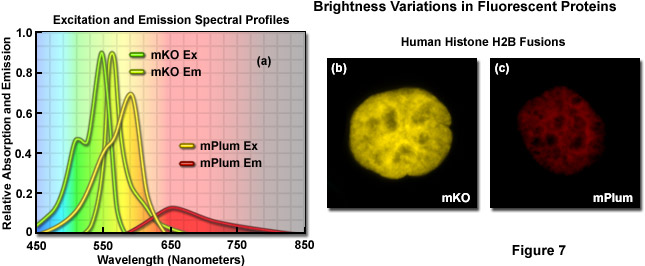
Illustrated in Figure 7 are variations in brightness between two fluorescent proteins derived from reef corals as revealed by imaging and examination of the spectral profiles. Kusabira Orange is a monomeric protein (abbreviated mKO) having an absorption maximum at 548 nanometers and an emission maximum of 559 nanometers. The molar extinction and quantum yield of mKO are 63,800 M-1 and 0.60, respectively, which render the fluorescent protein approximately 90 percent as bright as EGFP. In contrast, mPlum (Figure 7(c)) is only 10 percent as bright as EGFP due to a lower extinction coefficient (41,000M-1) and quantum yield (0.10). A comparison of the spectral profiles of mKO and mPlum are presented in Figure 7(a). Note the relatively small integrated intensity for the emission profile of mPlum compared to mKO, which is due to the differences in quantum yield. Images of the two fluorescent proteins fused to human histone H2B are presented in Figures 7(b) and 7(c). At similar expression levels, when the camera exposure time and gain are held constant, the image of mKO has significantly more intensity than that of mPlum. Thus, under similar conditions, images of cells expressing mKO can be expected to have greater signal-to-noise than those from cells expressing mPlum.
Fluorescent protein maturation efficiency is also determined by a number of variables, including the biological origin of the protein and the abundance of species-specific optimum codons in the organism chosen for expression. In general, fluorescent proteins that have been codon-optimized for expression in mammalian cells will be expressed with high efficiency and mature rapidly at 37° C, although a small percentage of the product may not fold correctly, resulting in poor targeting and high background fluorescence. Many of the protein variants derived from the original Aequorea jellyfish green fluorescent protein have been optimized (with regards to the wild-type GFP) through mutagenesis for expression in mammalian systems at high efficiency. Fluorescent proteins from reef corals and sea anemones generally express well at 37° C, presumably because the native species from which the proteins are obtained have evolved in warmer habitats. The presence of molecular oxygen is also a critical factor in fluorescent protein chromophore development during the maturation process. During the formation of chromophores in Aequorea protein variants, at least one oxygen molecule is required for dehydrogenation, while reef coral proteins that emit in the orange-red spectral regions usually require two molecules. In mammalian cell cultures, fluorescent protein expression is rarely hampered by a lack of oxygen, but anoxia could become a limiting factor in other systems.
Fluorescence Filter Combinations for Fluorescent Proteins
Regardless of the intrinsic brightness displayed by a particular fluorescent protein, the ability to achieve high signal levels in optical microscopy is primarily determined by the configuration of the imaging equipment. The line spectrum of a laser system or an arc-discharge plasma lamp coupled to fluorescence filters that are used to excite the fluorescent protein should strongly overlap the chromophore absorption profile. Emission filters should have the widest possible passband region coinciding with the emission spectrum of the fluorescent protein. In addition, the camera system must be capable of recording images with high quantum efficiency in the fluorescence emission region of interest, and the optical system of the microscope should have high throughput in the wavelength bands necessary for producing excitation and gathering emission. Even with research-level instrumentation, it is often quite difficult to achieve the maximum potential fluorescent protein brightness levels in each spectral class unless the fluorescence filter sets are completely optimized for imaging the proteins. Many core imaging facilities have limited inventories of filter sets that are typically designed for traditional synthetic fluorophores rather than fluorescent proteins. For example, the standard DAPI, FITC, TRITC, and Texas Red fluorescence filter combinations, which are often marketed by default with widefield arc-discharge microscopes, are not suitable for many fluorescent proteins and are less than optimal for others.
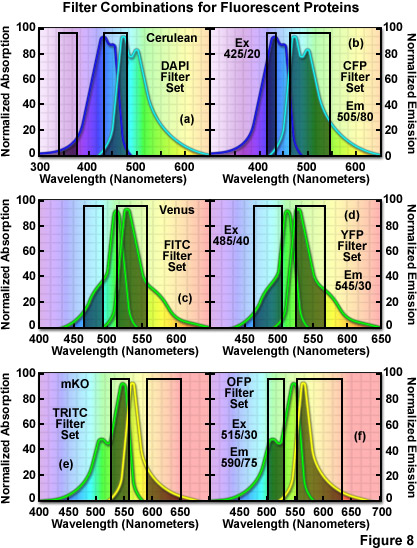
The hazards of using incorrect filter sets for imaging fluorescent proteins are illustrated in Figure 8 for several of the more useful fluorescent proteins with "standard" widefield microscopy filter sets. The cyan fluorescent protein, Cerulean, is depicted in Figure 8(a) and 8(b) with the absorption and emission spectral profiles superimposed over a DAPI filter set (Figure 8(a)) as well as a set designed specifically for cyan fluorescent proteins (Figure 8(b)). Note the poor overlap of the DAPI filters with the Cerulean absorption and emission bands, which yields low signal levels when this combination is employed. Narrowing the excitation band to 20 nanometers and moving the center wavelength 85 nanometers to longer wavelengths significantly improves the excitation of Cerulean (Figure 8(b)). Likewise, the emission filter effectively captures a majority of the fluorescence emission with a wideband (80-nanometer) filter having a center wavelength of 505 nanometers. In general, cyan fluorescent proteins are best imaged with custom filter sets specifically designed for their blue-violet excitation range.
The standard FITC filter set is a much closer match for the absorption profile of the yellow fluorescent protein, Venus (Figure 8(c)), but a better combination increases the excitation filter bandwidth to 40 nanometers and moves the center wavelength to 485 nanometers (red-shifted by 5 nanometers; Figure 8(d)). The optimum emission filter bandwidth for Venus is similar to the FITC version with an increase of 10 nanometers for the center wavelength. Most of the green and yellow fluorescent proteins can be satisfactorily imaged with the FITC filter set, but quantitative imaging benefits from optimizing the central wavelengths and bandwidths for each protein. Orange fluorescent proteins have spectral profiles that fall between the FITC and TRITC filter wavelength regions for optimal excitation and gathering of emission. Imaging mKusabira Orange with a TRITC filter combination (Figure 8(e)) is a good example of the mismatch. The TRITC excitation filter covers an acceptable cross-section of the absorption spectrum, but the emission bandpass region is shifted slightly too far towards the low-intensity longer emission wavelengths (Figure 8(e)). In addition, the emission filter for this set captures only a small portion of the fluorescent protein emission spectral "tail", leading to a loss of signal. A more effective filter set for mKusabira Orange is illustrated in Figure 8(f). Fortunately, a majority of the red fluorescent proteins (including DsRed, mCherry, JRed, tdTomato, and mPlum) can be imaged with good results using either the TRITC or Texas Red filter combinations.
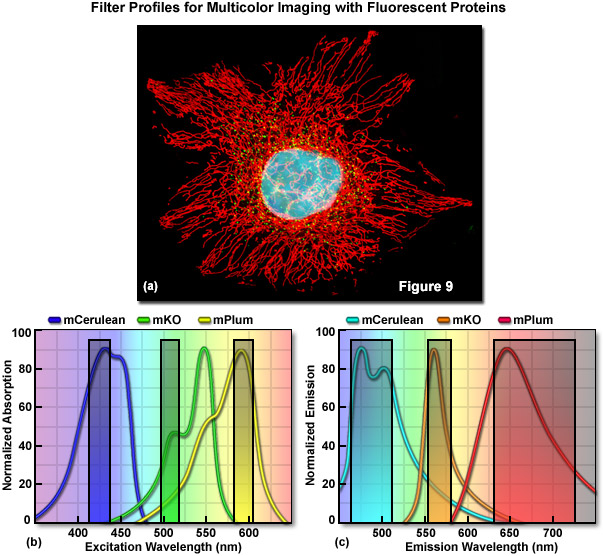
Optimized fluorescence filter combinations for multicolor imaging of three fluorescent proteins spanning the cyan to far-red wavelength regions are presented in Figure 9. In Figure 9(a) a widefield fluorescence image of a single HeLa cell labeled with mCerulean (human histone H2B; nucleus; pseudocolored cyan), mKusabira Orange (peroxisomes; pseudocolored green), and mPlum (mitochondrial targeting signal; pseudocolored red) is shown. Excitation filters optimized to avoid cross excitation for mCerulean, mKusabira Orange, and mPlum fluorescent proteins having center wavelengths of 425, 508, and 585 nanometers, respectively are outlined in Figure 9(b). The bandwidth of the excitation filters is 20 nanometers. In Figure 9(c), emission filter profiles are optimized for the same probes having center wavelengths of 480, 564, and 675 nanometers with bandwidths of 40, 28, and 100 nanometers, respectively.
Phototoxicity and Photostability
Aside from the toxicity that occurs due to excessive concentrations of synthetic fluorophores and the over-expression or aggregation of poorly localized fluorescent proteins, the health and longevity of optimally labeled mammalian cells in microscope imaging chambers can also suffer from a number of other deleterious factors. Foremost among these is the light-induced damage (phototoxicity) that occurs upon repeated exposure of fluorescently labeled cells to illumination from lasers and high-intensity arc-discharge lamps. In their excited state, fluorescent molecules tend to react with molecular oxygen to produce free radicals that can damage subcellular components and compromise the entire cell. In addition, several reports have suggested that particular constituents of standard culture media, including the vitamin riboflavin and the amino acid tryptophan, may also contribute to adverse light-induced effects on cultured cells. Fluorescent proteins, due to the fact that their fluorophores are buried deep within a protective polypeptide envelope, are generally not phototoxic to cells. In contrast, many of the common synthetic fluorophores, such as the MitoTracker and most nuclear stains (including Hoechst, SYTO cyanine dyes, and DRAQ5), can be highly toxic to cells when illuminated for even relatively short periods of time. In designing experiments, synthetic fluorophores as well as fluorescent proteins that exhibit the longest excitation wavelengths possible should be chosen in order to minimize damage to cells by short wavelength illumination. Thus, rather than creating fusion products with blue or cyan fluorescent proteins (excited by ultraviolet and blue illumination, respectively), variants that emit in the yellow, orange, and red regions of the spectrum should, when possible, be used instead.
Investigators should take care to perform the necessary control experiments when using new fluorescent protein variants, chimeric fusion protein vectors, and cell lines to ensure that cytotoxicity and phototoxicity artifacts do not obscure important biological phenomena. In some cases, lipophilic reagents induce deleterious effects that may be confused with fluorescent protein toxicity during imaging in cell lines following transient transfections. Oligomeric fluorescent proteins (discussed below) from reef corals have a far greater tendency to form aggregates (combined with poor subcellular localization) than do the monomeric or weakly dimeric jellyfish proteins, but improperly folded fusion products can occur with any variant. Recently, a fluorescent protein capable of generating reactive oxygen species (ROS) upon illumination with green light has been reported as an effective agent for inactivation of specific proteins by chromophore-assisted light inactivation (CALI) and photodynamic therapy. Appropriately named KillerRed, this genetically encoded photosensitizer is capable of killing both bacteria and eukaryotic cells upon illumination in the microscope. Previous studies on GFP phototoxicity indicate that even through the chromophore is capable of generating singlet oxygen, the probe is inefficient as a photosensitizer. However, prolonged illumination of cells expressing GFP and its variants can result in physiological alterations and eventual cell death, a definite signal of the potential for phototoxicity in long-term imaging experiments.
In live-cell experiments, fluorescent proteins are highly advantageous for extended imaging due to their reduced rate of photobleaching when compared to synthetic fluorophores. Although there is a high degree of uncorrelated variability between fluorescent proteins in terms of photostability, most variants are useful for short-term imaging (from 1 to 25 captures), while several of the more photoresistant proteins can be employed in time-lapse sequences that span periods of 24 hours or longer (in which hundreds to thousands of images are gathered). The long term stability of any particular protein, however, must be investigated for every illumination scenario (widefield, confocal, multiphoton, spinning disk, swept-field, etc.) because differences in photostability are often observed with the same protein when illumination is produced by an arc-discharge lamp versus a laser system. Thus, in terms of photostability, the selection of fluorescent proteins is dictated by numerous parameters, including the illumination conditions, the expression system, and the effectiveness of the imaging setup.
Fluorescent Protein Oligomerization
All of the fluorescent proteins discovered to date display at least a limited degree of quaternary structure, exemplified by the weak tendency of native Aequorea victoria green fluorescent protein and its derivatives to dimerize when immobilized at high concentrations. This tenant is verified by the obligate dimerization necessary for solubility in fluorescent proteins isolated from Renilla sea pansies and the almost universal tetramerization motif of the native yellow, orange, and red fluorescent proteins isolated in reef corals and anemones. Oligomerization can be a significant problem for many applications in cell biology, particularly in cases where the fluorescent protein is fused to a host protein that is targeted at a specific subcellular location. Once expressed, the formation of dimers and higher order oligomers induced by the fluorescent protein portion of the chimera can produce atypical localization, disrupt normal function, interfere with signaling cascades, or restrict the fusion product to aggregation within a specific organelle or the cytoplasm. This effect is particularly marked when the fluorescent protein is fused to partners such as actin, tubulin, connexins, or histones, which themselves participate in natural oligomer formation. Fusion products with proteins that form only weak dimers (in effect, most Aequorea victoria GFP variants) may not exhibit aggregation or improper targeting, provided the localized concentration remains low. However, when dimeric fluorescent proteins are targeted to specific cellular compartments, such as the plasma membrane, the localized protein concentration can, in some circumstances, become high enough to permit dimerization. This is a particular concern when conducting resonance energy transfer (FRET) experiments, which can yield complex data sets that are easily compromised by dimerization artifacts.
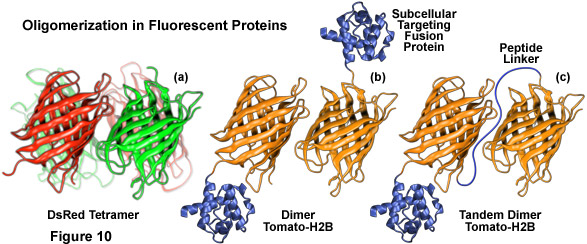
Illustrated in Figure 10 are several oligomerization motifs commonly observed in fluorescent proteins. The tetrameric DsRed fluorescent protein (Figure 10(a)) shows a high level of structural similarity with green fluorescent protein (see Figure 1) at the monomer level, but the intermolecular interactions are far more complex for DsRed. Two of the monomeric units in Figure 10(a) are shaded green to arbitrarily illustrate the fact that not all participating members in the DsRed tetramer mature fully to the red species. Figure 10(b) depicts the dimeric fluorescent protein Tomato fused to the amino acid sequence for human histone H2B, which participates in formation of nucleosomes. When coupled to a fluorescent protein that forms obligate dimers, histone H2B is severely compromised with regards to its ability to participate unhindered in chromosome condensation and cells expressing high levels of this fusion product usually do not survive.
The basic strategy for overcoming oligomerization artifacts is to modify the fluorescent protein amino acid sequence to include residues that disrupt intermolecular binding, a procedure that varies in complexity depending upon the nature and origin of the protein. For many of the Aequorea GFP variants, dimerization can be either significantly reduced or eliminated completely by replacing the hydrophobic amino acid side chains in the dimer interface with positively charged residues at several key sequence positions. The three most successful mutations, in ascending order of effectiveness, are F223R, L221K, and A206K, where the non-polar amino acids phenylalanine, leucine, and alanine are replaced by their hydrophilic relatives, arginine or lysine. In cases where close molecular associations are suspected involving a fusion protein and where quantitative FRET interactions are being investigated, it is highly recommended that Aequorea GFP variants (ECFP, EGFP, EYFP) should be converted into monomers using one of these point mutations.
Creating fluorescent protein monomers from the tetrameric coral reef and sea anemone proteins is usually far more difficult. For example, even at exceedingly low concentrations, the DsRed fluorescent protein is an obligate tetramer that cannot be dissociated without irreversible denaturation of the polypeptides. In the tetrameric unit, each DsRed protein interacts with two adjacent neighbors, one through a hydrophobic interface and the other through a hydrophilic interface resulting in a complex assembly (see Figure 10(a)). Other Anthozoa proteins, such as the Zoanthus variants and eqFP611, have simpler interfaces that may prove easier to disrupt into monomers. The most successful approaches utilized so far to generate fluorescent protein monomers with Anthozoa species have involved repeated site-directed mutagenesis to disrupt the tetrameric interfaces, usually by substitution of hydrophilic or charged amino acids for hydrophobic and neutral moieties. Because a significant decrease in fluorescence emission quantum yield usually accompanies these genetic modifications, a second round of random mutagenesis is often necessary to rescue fluorescence.
Another technique involves creating vectors containing two sequential coding regions separated by a short linker of non-specific amino acids, known as tandem dimers (Figure 10(c)), from proteins that exist naturally as dimeric and tetrameric complexes. Upon expression, the fused fluorescent proteins preferentially bind to each other to form an intramolecular dimeric unit that performs essentially as a monomer, although at twice the molecular weight (and size). This method was successfully applied with HcRed by fusing two copies of the DNA sequence, separated by a short linker of four amino acids, to several subcellular localization proteins. Tandem dimer constructs have also been developed with DsRed, dimeric Tomato (Figure 10(b)), and a photoconversion optical highlighter fluorescent protein known as Eos. Other mechanisms for reducing fluorescent protein oligomerization and aggregation effects include removing several basic residues from the N-terminus and simultaneous co-expression of fluorescently tagged proteins with an excess of either fusion partner alone or with a non-fluorescent mutant of the marker protein. Regardless of the specific mechanism employed to overcome fluorescent protein oligomerization, the most important point is that experimental results are not compromised by artifacts induced by the existence of quaternary structures.
Fluorescent Protein Fusion Chimeras
In order to adapt the jellyfish fluorescent proteins for use in mammalian systems, several basic amino acid modifications of the wild-type green fluorescent protein were undertaken and are now found in all commonly used variants. The first step was to optimize the development of fluorescence for a 37° C environment. Maturation of the wild-type fluorophore is quite efficient at 28° C, but increasing the temperature to 37° C substantially reduces maturation rate and results in decreased overall fluorescence. Mutation of the phenylalanine residue at position 64 to leucine (F64L) results in improved maturation of fluorescence at 37° C, which is at least equivalent to that observed at 28° C. This mutation is present in the most popular varieties of fluorescent proteins derived from Aequorea victoria, but is not the only mutation that improves folding at 37° C, as other variants have been discovered.
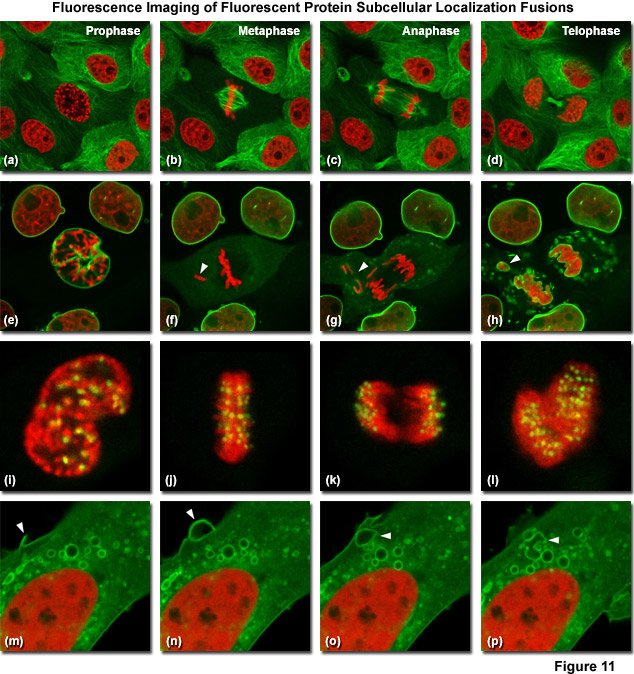
Illustrated in Figure 11 are fluorescent protein fusions to targeting proteins and peptides in action imaged with spinning disk confocal and widefield microscopy. The first four panels visualize mitosis in dual-labeled normal pig kidney (LLC-PK1 cell line) epithelial cells stably expressing fusions of mCherry-H2B (histones) and mEmerald-alpha-tubulin. In Figure 11(a), a cell in prophase is captured adjacent to several cells in interphase. The dividing cell forms a spindle and enters metaphase (Figure 11(b)), and during anaphase (Figure 11(c)), the spindle poles translocate to opposite sides of the cell, pulling the condensed chromosomes along. Finally, the chromosomes begin to decondense during telophase (Figure 11(d)) as the daughter cells recover from cell division (mid-body visible). Figures 11(e) through 11(h) present dispersion of the nuclear envelop during mitosis. HeLa cells expressing fusions of mRuby-H2B and mEmerald-lamin B1 are imaged undergoing mitosis. In Figure 11(e), the central cell is imaged in late prophase with the nuclear envelop intact. During metaphase (Figure 11(f)), the nuclear envelope signal is dispersed in the cytoplasm. Note the detached chromosome (arrow). During late anaphase (Figure 11(g)), the nuclear envelop begins to reform. Note the independent mitosis event for the detached chromosome (arrow). Telophase nuclei (Figure 11(h)) decondense and the nuclear envelope reforms. Note the separate nuclear envelope formation on the detached chromosomes (arrow). Figures 11(i) through 11(l) depict HeLa cells labeled with a fusion of mApple-H2B and mEmerald-CENPB undergoing mitosis. The prophase nucleus (Figure 11(i)) shows labeled condensed chromosomes (red) and centromeres (green). Centromeres and chromosomes are visible during cell division in metaphase (Figure 11(j)), anaphase (Figure 11(k)), and telophase (Figure 11(l)). In the bottom row of panels, (Figure 11(m) through Figure 11(p)) vesicle formation is observed in U2OS epithelial cells labeled with fusions of mEmerald-C-Src (membrane) and mRuby-H2B. Arrows denote formation of a vesicle at the periphery of the plasma membrane.
Aside from the improvement in chromophore maturation at 37° C, the optimization of codon usage for mammalian expression has also improved overall brightness of green fluorescent protein derivatives when expressed in mammalian cells. In all, over 190 silent mutations have been introduced into the coding sequence to enhance expression in human tissues. A Kozak translation initiation site (containing the nucleotide sequence A/GCCAT) was also introduced by insertion of valine as the second amino acid. These, along with a variety of other improvements, have resulted in a very useful cadre of fluorescence probes for live cell imaging of mammalian cells and are common to all of the currently used fluorescent variants derived from the original jellyfish protein.
The critical parameters involved in designing fluorescent protein chimera fusion products include the fluorescent protein coding sequence location within the chimera, the judicious use of amino acid linker sequences as spacers between the fluorescent protein and the fusion product, the promoter characteristics, and mechanisms to test the fidelity of the final product. In general, most fusions designed for subcellular localization place the fluorescent protein sequence on either the N or C terminus of the target protein (although fusions to circularly permuted fluorescent proteins are becoming more common). Investigators should first examine structure and function data (if available) to determine the most appropriate location for the fluorescent protein. For example, in fusions to a protein that must interact with a partner near one terminus, the opposite end of the protein would be the best candidate for hosting a fluorescent protein. In many cases, similar fusion products that have been successful with related proteins or in other species can be used as a guide for designing new chimeras. Absent previous data, however, the investigator is left only with empirical guesswork about the most ideal location for fluorescent protein and must often construct fusion products at both termini.
Instead of fusing the fluorescent protein sequence directly to that of the partner host protein, an intervening short segment of non-functional amino acids is typically added, both as a convenience in cloning technique as well as conduit to increase the molecular mobility at the junction. Linker sequences, in the ideal case, should be composed of amino acids having simple side chains, such as glycine and alanine, whose structures allow maximum flexibility, although serine residues are often interspersed into longer chains to enhance the solubility of polyglycine and polyalanine domains. In practice, however, successful fusion products have been generated that contain a wide spectrum of intervening spacer configurations, ranging from no linker amino acids to 20 or more. Often, especially when using commercial cloning vectors, the use of multiple cloning sites (MCS) to enable convenient in-frame insertions results in charged or hydrophobic amino acid residues being incorporated into the fusion product. In the final analysis, the choice of linker is often very flexible and a variety of combinations may ultimately produce useful chimeras. Adding a few extra amino acids between the fluorescent protein and the host target usually does not affect expression or localization, although reports of linker design being a critical aspect of chimera success do exist.
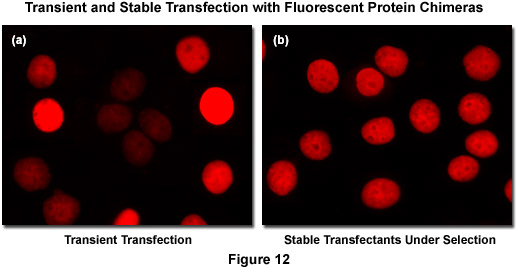
Recombinant DNA containing the nucleotide coding sequences for fluorescent proteins, either alone or fused to subcellular localization polypeptides (usually termed a vector), is readily introduced into mammalian (or other eukaryotic) cells by a process known as transfection. During the transfection procedure, transient gaps are chemically or physically created in the cell membrane that enable external macromolecules, such as supercoiled plasmid DNA, to enter the cytoplasm and be expressed by the cell's protein synthesizing machinery. Because transfected viral or plasmid DNA is usually not permanently incorporated into the mammalian cell genome and is only expressed temporarily, transfection is dissimilar from the process of transformation where the foreign DNA genetically alters the cell. However, specific vectors have been created to exploit a variety of cell characteristics and these agents are often suitable for producing modified cell lines that incorporate at least a portion of the extraneous DNA into the genome, thus being able to continually express the target protein over many generations. Transfections of this nature are termed stable (Figure 12(b)) whereas temporary transfections are referred to as transient (Figure 12(a)). In transient fluorescent protein transfections, expression levels are highly varied, whereas taking more time to select stably-expressing cell populations often produces superior results. It is often tempting (and much quicker) to transiently express the fusion and search for cells exhibiting low levels of fluorescence intensity that may correspond to expression levels minute enough as to not interfere with normal function (see Figure 12(a)). However, producing stable cell lines presents the opportunity for a quantitative comparison of fusion expression to that of the endogenous protein and is a much safer bet. Alternatively, placing the fusion construct into a vector having an inducible promoter enables control in modulating the level of expression.
Conclusions
The current thrust of fluorescent protein development strategies is centered on fine-tuning the current palette of blue to yellow variants derived from the Aequorea victoria jellyfish, while simultaneously developing monomeric fluorescent proteins emitting in the orange to far-red regions of the visible light spectrum. Progress toward these goals has been substantial, and it is not inconceivable that near-infrared emitting fluorescent proteins loom on the horizon. The latest efforts in jellyfish variants have resulted in new and improved monomeric probes for the blue, cyan, green, and yellow regions, while the relentless search for a bright monomeric and fast-maturing red fluorescent protein has yielded a host of excellent candidates spanning the longer wavelengths. Continuing efforts in protein engineering of the existing fluorescent proteins, coupled with advanced new technologies, should further expand the color palette and ultimately provide proteins in every spectral class that mature rapidly and are bright and photostable.
Contributing Authors
David W. Piston - Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, Tennessee, 37232
Robert E. Campbell - Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada, T6G2G2
Richard N. Day - Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Dr., Indianapolis, Indiana, 46202.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.