







Introduction
Efforts to identify the molecular basis for the novel fluorescence in jellyfish began with Osamu Shimomuras studies of the Aequorea jellyfish in the early 1960s. Located then at Princeton University, Shimomura traveled with Frank Johnson to Friday Harbor Laboratories at the University of Washington with the goal of developing a method to extract the light emitting components from the jellyfish, Aequorea victoria. The investigators observed that when the circumoral ring on the underside of the jellyfish, which contains the luminescent organs, was removed and squeezed through a sieve, the squeezate was dimly luminescent. In a heroic effort, involving the collection and processing of many thousands of jellyfish into squeezate, they isolated and purified a chemiluminescent protein that was subsequently named aequorin.
Aequorin is a luciferase that catalyzes the oxidization of the substrate coelenterazine in a calcium-dependent reaction that leads to the emission of blue light. This discovery was curious, however, since the luminescent organs of the intact jellyfish produce green light. Importantly, during the purification Shimomura had observed strong green fluorescence under ultraviolet illumination, demonstrating that there was an autofluorescent protein in the jellyfish extracts. It was later determined that this autofluorescent protein was a companion protein for the chemiluminescent aequorin - the protein now known as the jellyfish green fluorescent protein (GFP). In the 1970s, Shimomura and colleagues purified this autofluorescent protein and showed for the first time that GFP, via an energy transfer process, absorbed the excited state energy from aequorin and emitted green light.
The identification of GFP from Aequorea was the first step in what has often been described by many as a revolution in cell biology, although it would be some years before the true significance of this observation became apparent. Early in the 1980s, while working in collaboration with Milton Cormier at the University of Georgia, Douglas Prasher prepared a cDNA library from Aequorea victoria mRNA, and used this to clone the gene encoding the aequorin. Later, using the same cDNA library, Prasher isolated another clone that contained part of the sequence encoding the companion protein, GFP (pGFP1). Later, by creating a new cDNA library in the lgt10 cloning vector, and screening it with the pGFP1 clone, Prasher obtained a single full-length clone (gfp10) that encoded the complete Aequorea GFP sequence in 1992. Prasher recognized the great potential of GFP as a tool for cell biologists, but since no information was available regarding the biosynthesis pathway leading to chromophore formation and fluorescence, he was cautious of the difficulties associated with producing the protein in biological systems other than the jellyfish.
Fortunately, Douglas Prashers concerns regarding the difficulties of expressing Aequorea GFP in other biological systems ultimately proved to be unwarranted. Using the clone isolated by Prasher, Martin Chalfie successfully expressed GFP in prokaryotes (bacteria) and the sensory neurons of Caenorhabditis elegans nematode worms, demonstrating for the first time that chromophore formation (and fluorescence) required no additional factors that were specific to the jellyfish. A wide variety of other studies rapidly followed, which demonstrated the utility of this new genetically-encoded probe for in vivo fluorescence labeling in a variety of different cellular systems and transgenic organisms.
As it turns out, the GFP chromophore is encoded by the primary amino acid sequence, and forms spontaneously without the requirement for cofactors or external enzyme components (other than molecular oxygen), through a self-catalyzed protein folding mechanism and intramolecular rearrangement. Thus, genetically-encoded GFP provided for the first time the ability to label specific proteins inside the living cell without the need for exogenous synthetic or antibody-labeled fluorescent tags. When coupled to the astonishing advances in live-cell imaging instrument technologies that have occurred over the past decade, the fluorescent proteins have truly ushered in a new era in studies of cell biology and physiology. These tools now provide an important complement to the classical biochemical studies, extending the analysis of protein-protein interactions, protein conformational changes, and the behavior of signaling molecules to their natural environment within the intact cell.
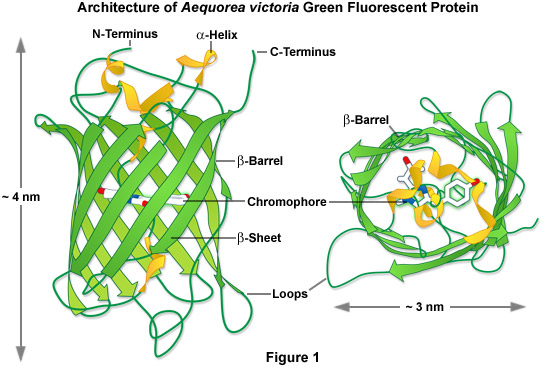
In one of the first investigations of jellyfish bioluminescence by Shimomura using the purified Aequorea victoria GFP, it was demonstrated that the entire protein sequence was necessary for its characteristic fluorescence (see Figure 1). However, the digestion of the purified protein with papain, followed by high-performance liquid chromatography to isolate the fragments, revealed that a single hexapeptide sequence starting at amino acid 64 was responsible for all the light absorption properties of GFP. This discovery led to the demonstration that the chromophore absorbing the blue light energy from aequorin was formed by the cyclization of the adjacent serine(65)-dehydrotryrosine(66)-glycine(67; the number denotes the amino acid position in the intact peptide sequence) residues within this hexapeptide sequence.
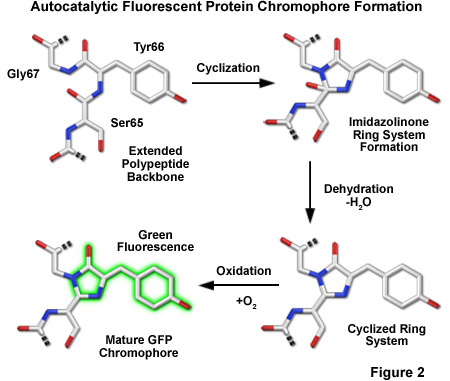
The most popular model describing the formation of the chromophore during the maturation of GFP has a series of torsional peptide and side-chain bond adjustments that position the carboxyl carbon of Ser65 close to the amino nitrogen atom of Gly67 (Figure 2). This leads to a nucleophilic attack by this carbon atom on the amide nitrogen of glycine, which is followed by dehydration, resulting in the formation of an imidazolin-5-one heterocyclic ring system. Fluorescence occurs when oxidization of the Tyr66 alpha-beta carbon bond by molecular oxygen extends the conjugation of electron orbitals of the imidazoline ring system to include the tyrosine phenyl ring, and its para-oxygen substituent. The participation of molecular oxygen is a critical factor in chromophore development, and at least one oxygen molecule is required for dehydrogenation. Additionally, Arg96 and Glu222, two residues that are invariant in every fluorescent protein yet discovered, are thought to catalyze the reaction. The end result of chromophore maturation is a highly conjugated π-electron resonance system that largely accounts for the spectroscopic and photophysical properties of the protein.
In 1996, the crystal structure for GFP was solved, revealing that the cyclic tripeptide chromophore is buried in the center of a nearly perfect cylinder formed by a tightly interwoven eleven-stranded beta-barrel structure (see Figure 1). As the protein folds, the tripeptide sequence is positioned at the core of the beta-barrel, driving the cyclization and dehydration reactions necessary to form the mature chromophore (illustrated in Figure 2). Visualization of the intact structure of GFP explained the earlier observations that nearly the entire protein sequence is required to generate a functional chromophore. The remarkable cylindrical geometry of GFP is conserved in all the fluorescent proteins yet discovered, and appears to be ideally suited to the primary function of protecting the chromophore.
The beta-barrel structure has dimensions of approximately 30 by 40 Angstroms, and the tight packing of amino acid residues imparts a high level of stability to the protein. A lack of clefts and gaps for access of small ligands (such as ions and oxygen), combined with the fact that the chromophore is located near the exact center of the protein (almost perpendicular to the long axis of the barrel), partially explains the extraordinary photostability and high quantum yields that are observed. Fortunately for the cell biologists, both the amino and carboxy termini are exposed on the surface of the barrel, and are therefore available to be employed as linkers to fusion proteins without significantly affecting the structural integrity of the fluorophore. The compact protein structure also enhances resistance to changes in pH, temperature, fixation with paraformaldehyde, and the disruptive action of many common denaturing agents, such as urea and guanidinium hydrochloride.
back to top ^Aequorea GFP Spectral Variants
Over the last decade, the application of both site-directed and random mutagenesis approaches to the cDNA encoding the Aequorea-GFP demonstrated that its fluorescence properties are very dependent on the arrangement and three-dimensional structure of amino acid residues surrounding the chromophore. As will be discussed in detail below, mutations that alter the residues immediately adjacent to the chromophore generally have a significant impact on the spectral properties of the protein. Surprisingly, several amino acid substitutions in regions of the polypeptide far removed from the chromophore were also found to profoundly affect the spectral characteristics of the protein. These protein-engineering approaches have generated a number of different spectral variants from Aequorea-GFP, and some of the more significant derivatives will be described in the following sections. Just as important, site-directed mutagenesis was also used to introduce substitutions that improved the efficiency of protein maturation and expression in many different heterologous cell systems. For example, preferred human codon usage was incorporated for improved expression in mammalian cells, as well as silent mutations that increased the efficiency of folding and maturation of the protein at physiological temperatures. These variants are collectively termed the enhanced (E) fluorescent proteins.
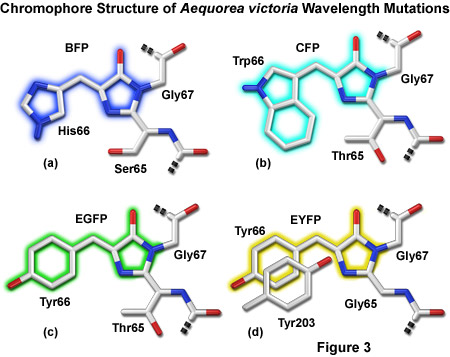
Presented in Figure 3 are representatives of the four major color classes of Aequorea-GFP derivatives. Substitution of histidine for tyrosine in EGFP yields an enhanced blue variant (see below) shown in Figure 3(a). Cyan derivatives contain a tryptophan residue in place of Tyr66, as depicted in Figure 3(b). Note that the Trp66 side chain is illustrated in the cis conformation as occurs for Cerulean derivatives rather than the trans isomer that is common to many cyan variants of Aequorea-GFP. Substitution of Ser65 for threonine (Figure 3(c)) produces the "enhanced" green derivative that will be discussed in a following section. Stacking of a second tyrosine (Tyr203) on Tyr66 along with substitution of Ser65 for glycine yields Aequorea derivatives emitting in the yellow-green region. In Figure 3, portions of the chromophores that are conjugated and give rise to fluorescence are shaded with colors corresponding to the emission spectral profile.
back to top ^Green Fluorescent Proteins
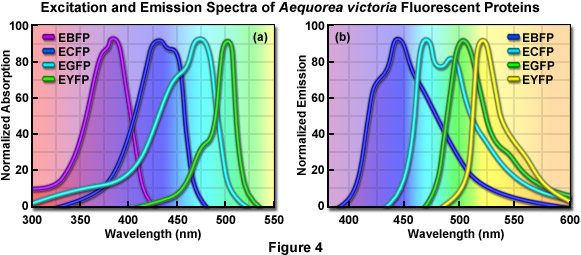
The wild type (wt) Aequorea GFP displays a complex absorption spectrum, with maximal excitation occurring at 397 nanometers, and a minor secondary peak of residing at 476 nanometers. A number of investigators have examined the dynamics of wtGFP protein in the excited state, and showed that fluorescence resulted from deprotonation of the Tyr66 residue within the chromophore that resulted in an ionic species favoring excitation at 476 nanometers. The predominate ground state species contains a protonated Tyr66 residue and is responsible for the larger 397 nanometers absorption peak. In addition, it is thought that the charged intermediate state exists as a minor population of the ground (non-excited) state, accounting for the minor secondary absorption peak at 476 nanometers. The complex absorption spectrum featuring a significantly higher extinction coefficient at near-ultraviolet wavelengths, coupled with the low quantum yield of wtGFP, have severely limited its utility for cellular imaging applications.
Mutagenesis strategies were initially applied to the sequence encoding the wtGFP in order to determine whether different amino acid substitutions might be used to fine-tune its spectral characteristics. This approach has yielded a broad range of derivative fluorescent proteins with fluorescence emission ranging from the blue to the yellow regions of the visible spectrum (see Table 1, Figure 3, and Figure 4). For example, an interesting variant of wtGFP, named Sapphire, resulted from substitution of the isoleucine for threonine at position 203 (T203I). This residue is located in one of the beta-sheet strands that surround a central alpha-helix containing the chromophore, and is positioned close to the chromophore, where it influences the local environment. The consequence of the T203I substitution is the loss the secondary absorption peak of GFP at 475 nanometers, resulting in an fluorescent protein exhibiting peak absorption at 399 nanometers and emission in the green spectral region (511 nanometers). These rather unusual spectral properties endow Sapphire with a Stokes shift of over 100 nanometers, one of the largest for the current cadre of fluorescent proteins. Several improved Sapphire variants were subsequently developed with more efficient maturation, including a probe known as T-Sapphire (T is an acronym for turbo). In addition, a yellow fluorescent protein having a long stokes shift (excitation at 406 nanometers and emission at 526 nanometers) has been developed. Named Ametrine, this protein also has found utility as a FRET donor for orange and red fluorescent proteins.
Early studies of the structure-function relationships in the GFP chromophore region by Roger Tsiens laboratory showed that mutations altering the first amino acid in the chromophore, Ser65, to cysteine, leucine, alanine, or threonine simplified the excitation spectrum to a single peak ranging from 471 to 489 nanometers. For example, changing the Ser65 to threonine (the S65T mutation) stabilized the hydrogen-bonding network in the chromophore, resulting in a permanently ionized form of the fluorophore absorbing at 489 nanometers. The resulting GFP-S65T mutant was a distinct improvement over wtGFP for applications as a fluorescent marker in living cells because it had a well-defined absorption profile with a single peak at 489 nanometers. In addition, the GFP-S65T derivative is about five-fold brighter than wtGFP, and it matures more rapidly, allowing fluorescence to be detected at earlier time points after cell transfection. The GFP-S65T variant was further modified by replacing phenylalanine for leucine at position 64 (F64L), which improved the efficiency of protein maturation at 37° C, yielding EGFP (enhanced GFP). This enhanced variant features an excitation spectral profile (see Figure 4(a)) that overlays nicely with the 488-nanometer argon-ion laser line and is similar in profile to fluorescein and related synthetic fluorophores that are readily imaged using commonly available filter sets designed for fluorescein (FITC). Furthermore, EGFP is among the brightest and most photostable of the Aequorea-based fluorescent proteins. The only drawbacks to the use of EGFP as a fusion tag are a slight sensitivity to pH and a weak tendency to dimerize.
Continued engineering of EGFP has yielded several additional green variants with improved characteristics. Among the best of these is the derivative called Emerald, which has improved photostability and brightness. Until recently, there was no commercial source for plasmids encoding Emerald (it is now available from Invitrogen), so there has been limited use of this high-performance green variant. Emerald fluorescent protein contains the S65T, and F64L mutations featured in EGFP, but also has four additional point mutations that further improve the efficiency of maturation and folding at 37° C, and increase the intrinsic brightness. Although Emerald is more efficient than EGFP in folding and developing fluorescence in mammalian cells, it has a fast photobleaching component that might affect quantitative imaging in some environments. Addition of the monomerizing A206K mutation, which is applicable to virtually all Aequorea GFP derivatives, may reduce or eliminate the fast photobleaching component in Emerald.
Among the most interesting new developments in the Aequorea GFP palette over the past several years is superfolder GFP (Table 1), which was designed to be capable of folding even when fused to insoluble proteins, as well as being slightly brighter and more acid resistant than either EGFP or Emerald. Investigators engineered superfolder GFP by fusing libraries of shuffled GFP sequences to a bait polypeptide that is relatively insoluble and usually interferes with GFP folding when expressed in bacteria. Starting with cycle-3 GFP, the EGFP mutations F64L and S65T were added and after four rounds of DNA shuffling, the investigators isolated a brightly fluorescent fusion clone (superfolder GFP) that contained six new mutations in addition to the cycle-3 and EGFP mutations. The work with superfolder GFP is clear evidence that there is significantly more room for engineering improvements even in the highly optimized GFP derivatives.
back to top ^Blue Fluorescent Proteins
One of the earliest color variants derived from the wtGFP was a blue fluorescent protein (BFP), which contains the substitution of Tyr66 with histidine (Y66H). The Y66H mutation produced a chromophore having a broad absorption band centered close to 380 nanometers, with blue light emission peaking at 448 nanometers (Figure 4). The original version of BFP had a low quantum yield and exhibited only about 15 to 20 percent of the brightness of the parent wtGFP, so there was clearly a need for additional mutations to improve the characteristics of the fluorescent protein. The introduction of these mutations led to the EBFP variant, but this derivative was still only 25 percent as bright as EGFP and its use as a cellular marker was severely limited by poor photostability.
In the mid-to-late 1990s, there was a keen interest in creating matched pairs of fluorescent proteins for fluorescence resonance energy transfer (FRET) experiments, as well as investigations requiring multicolor labeling. Because the fluorescence emission profile of BFP is readily distinguishable from EGFP, this fluorescent protein combination was the first utilized for dual color imaging. BFP also has the distinction of being incorporated into the first genetically encoded biosensor reporter proteins as a direct fusion to EGFP. The broad emission peak of BFP overlaps significantly with the absorption band of EGFP, allowing this pair to be used in FRET microscopy. The linkage of EBFP to EGFP through an intervening protease-sensitive spacer allowed FRET to be used to demonstrate the cleavage of the biosensor protein. In addition, BFP was used in combination with different GFP derivatives in FRET experiments designed to monitor transcription factor dimerization, calcium fluctuations, and apoptosis.
Properties of Selected Aequorea victoria Fluorescent Protein Variants
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 1
A compilation of properties of the most useful Aequorea-based fluorescent protein variants is presented in Table 1. Along with the common name and/or acronym for each fluorescent protein, the peak excitation (Ex) and emission (Em) wavelengths, molar extinction coefficient (EC), quantum yield (QY), relative brightness, photostability, and physiologically relevant quaternary structure are listed (* signifies a weak dimer). The computed brightness values were derived from the product of the molar extinction coefficient and quantum yield, divided by the value for EGFP. This listing was created from scientific and commercial literature resources and is not intended to be comprehensive, but instead represents fluorescent protein derivatives that have received considerable attention in the literature and may prove valuable in research efforts. The excitation and emission peak values listed may vary in published reports due to the broad spectral profiles. In actual fluorescence microscopy investigations, the experimental brightness of a particular fluorescent protein may differ (in relative terms) from the brightness provided in this table. Among the many potential reasons for these differences are wavelength-dependent variations in the transmission or reflectance of microscope optics and the efficiency of the camera. Furthermore, the extent of fluorescent protein folding and maturation will depend on both the particular variant being used as well as the characteristics and localization of the fusion partner.
Aside from its low intrinsic brightness and sensitivity to photobleaching, the utility of BFP for cellular imaging is also limited by its requirement for excitation with near-ultraviolet light, which is phototoxic to mammalian cells, even under limited illumination. Furthermore, substantially greater levels of cellular autofluorescence and light scattering are observed when exciting fluorophores in this spectral region, and microscope systems require specialized light sources, optics, and filter combinations for imaging at near-ultraviolet wavelengths. Hence, there was limited interest in developing more efficient EBFPs for almost a decade. Recently, however, several groups used advanced mutagenesis strategies to develop new BFP variants with much higher quantum yields and photostabilities, greatly improving the utility of these deep blue probes. These variants are named Azurite, SBFP2 (strongly enhanced blue fluorescent protein), and EBFP2. The most promising derivative, EBFP2, is the brightest and most photostable of the BFP variants yet discovered and was shown to be an excellent donor for FRET studies. EBFP2 should be useful for long-term imaging using living cells in situations where a blue probe is required, especially when two-photon excitation is used, which avoids cellular damage by near-ultraviolet excitation. All BFP variants can be readily imaged using standard BFP and DAPI fluorescence filter sets.
Substitution of Tyr66 with phenylalanine in wtGFP also produces a blue fluorescent protein, but in this case the spectral profiles are shifted to even shorter wavelengths than are observed with histidine at the same position (excitation at 360 nanometers and emission at 442 nanometers), and the resulting variant is extremely dim. Renewed interest in this chromophore was initiated with the introduction of an advanced derivative named Sirius that is much brighter and more photostable than EBFP2, as well as extremely insensitive to pH fluctuations (Table 1). Although Sirius is only approximately 10 percent as bright as EGFP, it features the shortest emission wavelength yet reported among fluorescent proteins (424 nanometers). Sirius was demonstrated to be stable at pH values ranging from 3 to 9 and exhibits potential as a FRET donor for cyan fluorescent proteins, but the requirement for excitation in the ultraviolet region (355 nanometers) will hinder attempts at long-term time-lapse imaging of live cells due to the typical increases in phototoxicity and autofluorescence often exhibited by probes in this spectral region, as discussed above.
back to top ^Cyan Fluorescent Proteins
The development of cyan (CFP) color variants from the Aequorea GFP provided an early alternative to the BFPs. Cyan fluorescence results from substitution of Tyr66 with tryptophan (T66W), and an enhanced version (ECFP) was generated by including several additional substitutions within the surrounding beta-barrel structure. The original T66W mutation yielded a chromophore featuring a broad, bimodal absorption spectrum that peaks at 433 and 445 nanometers, and an equally broad, bimodal fluorescence emission profile with maxima at 475 and 503 nanometers (Figure 4 and Table 1). Subsequent refinements, including the addition of the F64L substitution that improves maturation, and the S65T mutation (discussed above), resulted in an enhanced version (ECFP) with improved brightness and photostability. Even with these modifications, however, the brightness of ECFP is still only about 40 percent that of EGFP. Additionally, as was observed for the wtGFP, the complex excitation spectrum of ECFP indicates that there is more than one excited-state species for ECFP, which has been subsequently confirmed using fluorescence lifetime measurements. This attribute has limited the usefulness of ECFP as a probe for fluorescence lifetime imaging microscopy (FLIM), either alone or in combination with FRET investigations.
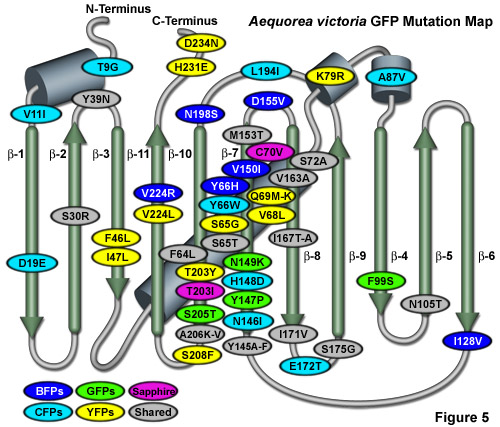
Illustrated in Figure 5 is an Aequorea victoria GFP mutation map showing many of the common mutations superimposed on a topological layout of the peptide structure. beta-sheets are numbered and depicted as thin, green cylinders with an arrow pointing toward the C-terminus, whereas alpha-helices are depicted by blue barrels. Mutations are color-coded to represent the variants to which they apply: BFPs (blue), CFPs (cyan), GFPs (green), YFPs (yellow), and Sapphire (violet). Folding, shared, and monomerizing mutations are indicated with gray ellipses. Note that almost 75 percent of the mutations are located in the central helix and beta-sheet strands 7, 8, and 10. In general, wavelength-specific mutations occur near the central helix containing the chromophore, while folding mutations occur throughout the sequence. Many of the cyan and yellow fluorescent protein mutations introduced near the ends of the protein resulted during the CyPet and YPet mutagenesis efforts discussed below. Several of the superfolder GFP folding mutations (S30R, Y39N, F99S, and N105T) also occur away from the chromophore. The monomerizing mutation, A206K, is useful for all known GFP derivatives, but is replaced by A206V in superfolder GFP and EBFP2.
Efforts to address the complex excited state characteristics of ECFP have yielded a high performance variant, termed Cerulean (after the sky-blue color), which resulted from targeted substitutions on the solvent-exposed surface of ECFP (Tyr145 and HIs148). Cerulean has a higher extinction coefficient, increased quantum yield, and is reported to have a simplified excited state when compared to ECFP (see Table 1). In addition, Cerulean is at least 1.5-fold brighter than ECFP, and demonstrates increased contrast and signal-to-noise when coupled with yellow-emitting fluorescent proteins, such as Venus, in FRET investigations. Indeed, the combination of Cerulean and one of the high-performance yellow-emitting Aequorea fluorescent proteins (Venus and Citrine) is currently the most popular pairing for FRET-based measurements. The advantageous features afforded by Cerulean make this protein one of the most useful Aequorea-based derivatives. However, when Cerulean is expressed in living cells, its fluorescence decay kinetics still indicate the presence of more than one excited state species. Techniques that measure the fluorescence lifetime of a fluorophore are most accurate when the probe has simple decay kinetics.
The introduction of beneficial folding mutations into ECFP resulted in new monomeric variants featuring enhanced brightness, solubility, and improved performance for FRET-based imaging approaches. Termed super CFPs (SCFPs), the engineered variants are significantly brighter than the parent protein when expressed in bacteria and almost two-fold brighter in mammalian cells. Investigators speculate that these high-performance fluorescent proteins should have improved utility for labeling cellular proteins, and may also be useful for creating new CFP-based FRET biosensors with an improved dynamic range. The optimal detection of CFPs using widefield fluorescence microscopy requires a specialized filter set that is available from the microscope companies or aftermarket filter manufacturers. When the CFPs are used for laser scanning confocal microscopy, the most efficient excitation is achieved with a 440-nanometer diode laser, but the more commonly available 405-nanometer and 458-nanometer laser lines can also be employed, albeit with significantly reduced excitation efficiency.
back to top ^Yellow Fluorescent Proteins
The longest-wavelength emitting variants of Aequorea GFP were generated after careful inspection of the native GFP crystal structure. The X-ray studies indicated that the Thr203 residue in the beta-barrel lies in close proximity to the chromophore, and the Sapphire mutation (T203I; discussed above) had already demonstrated that substitutions at this position had the potential to change the spectral profile of the protein. The targeted substitution of Thr203 with tyrosine (T203Y) was expected to induce π-orbital stacking, leading to the stabilization of the chromophore excited state dipole moment. The T203Y substitution (originally named mutant 10C) resulted in almost a 20-nanometer shift to longer wavelengths for both the excitation and emission spectra, generating a new fluorescent protein with yellowish-green emission (YFP). The enhanced version, EYFP, has become one of the brightest and most widely utilized of the fluorescent proteins (Table 1 and Figure 4). Unfortunately, EYFP is very sensitive to acidic pH, losing approximately 50 percent of its fluorescence at pH 6.5. In addition, EYFP is also very sensitive to chloride ions, and exhibits poor photostability when compared to many of the other Aequorea fluorescent protein variants. Nevertheless, several investigators have exploited the environmental sensitivity of YFP to develop biosensors that measure cytoplasmic pH and chloride ion concentrations.
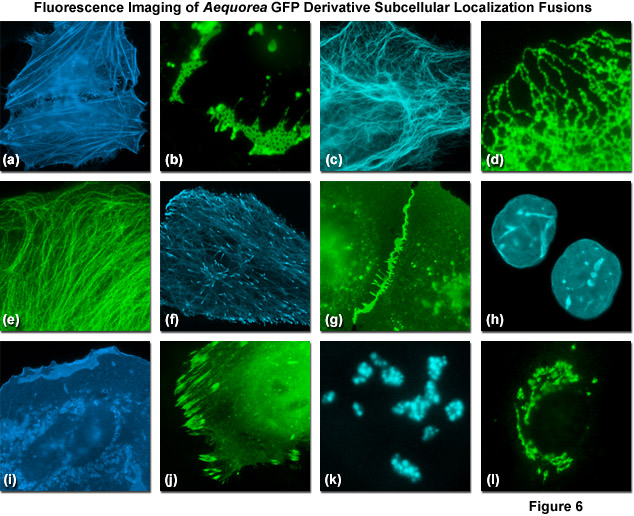
Presented in Figure 6 are fluorescence images captured in laser scanning confocal and spinning disk microscopy of Aequorea-GFP derivatives fused to subcellular localization targets. A fusion of EBFP2 to the actin-binding peptide domain known as Lifeact is shown highlighting the filamentous actin network in Figure 6(a). Figures 6(b) through 6(d) illustrate mVenus, mTurquoise, and mEmerald fluorescent proteins fused to connexin Cx43, cytokeratin, and an endoplasmic reticulum targeting signal, respectively. Figures 6(e) through 6(h) depict mEmerald, ECFP, mEGFP, and mCerulean3 fused to alpha-tubulin, microtubule end binding protein EB3, vascular epithelial cadherin, and lamin B1, respectively. Finally, Figures 6(i) through 6(l) show EBFP2, EGFP, mTurquoise, and mCitrine fused to C-Src, paxillin, fibrillarin, and a Golgi complex targeting sequence. All of the Aequorea-GFP derivatives perform quite well in most fusions, especially when compared to synthetic fluorophores or fluorescent proteins derived from reef corals and sea anemones.
Continued efforts to improve the YFP family led to the discovery that substitution of the glutamine at position 69 for methionine (Q69M) dramatically increases the acid stability of the protein, while simultaneously reducing its chloride sensitivity. This variant has been named Citrine in recognition of the yellow color and acid resistance. In addition, Citrine is expressed more efficiently in mammalian cell culture (especially when targeted to acidic organelles) and is more photostable than many previous yellow fluorescent proteins. Citrine features absorption and fluorescence emission maxima at 516 and 529 nanometers, respectively, and is 75 percent brighter than EGFP, although it is still much less photostable (Table 1). During continued studies, researchers demonstrated that substitution of the phenylalanine at position 46 with leucine (F46L) dramatically improved the maturation efficiency and reduced the halide sensitivity of YFP to yield a derivative that was named Venus (after the brightest object in the nighttime sky). Additional mutations were introduced that increased the tolerance of Venus to acidic environments. However, the photostability of Venus is only about 25 percent that of EGFP, which is a significant problem for long-term imaging experiments. Most of the Aequorea-based YFP derivatives (EYFP, Citrine, and Venus) are not available from commercial sources. A similar, but less well-characterized Aequorea derivative, named after the birthstone Topaz, is currently available from Invitrogen.
Several other EYFP variants have been introduced and may be useful for specialized applications. For example, the comprehensive site-directed mutagenesis strategies that yielded optimized monomeric variants of ECFP (discussed above) were also applied to EYFP to select for derivatives having enhanced brightness, folding efficiency, solubility, and utility in FRET applications. These efforts resulted in what has been termed a super yellow fluorescent protein derivative, SYFP, which is significantly brighter than the parent protein when expressed in bacteria and mammalian cells. Another potentially useful variant of EYFP was generated using an evolutionary mutagenesis strategy that was applied to cDNAs encoding a fused CFPYFP pair. Here, the goal was to simultaneously mutate CFP and YFP, and select for pairs with improved FRET efficiency. The cDNA libraries were screened directly for FRET efficiency and the best clones were subjected to several evolutionary cycles of random mutagenesis and synthetic DNA shuffling. This resulted in the generation of a new CFP-YFP pair, named CyPet-YPet (for Cyan or Yellow fluorescent Protein for energy transfer) that displayed a four-fold improvement in the ratiometric FRET signal.
A total of seven mutations were accumulated in CFP during the directed evolution to yield CyPet, which features absorption and emission maxima positioned at 435 nanometers and 477 nanometers, respectively. However, CyPet is only two-thirds as bright as Cerulean, and is not optimally expressed in cells grown at 37 °C, limiting it use for stand-alone applications. In contrast, YPet is the brightest of the YFP variants yet developed and demonstrates excellent photostability. In addition, YPet is more resistant to acidic environments than other YFP derivatives, which will enhance its utility in biosensor combinations targeted at acidic organelles. However, although the CyPet-YPet pair was selected as an optimized pairing for FRET studies, the poor quantum yield of CyPet has raised doubts about the origin of the improved FRET response. Recently, it was shown that the enhanced FRET signals resulted from S208F and V224L mutations that acted to stabilize an intramolecular complex formed between the linked CyPet and YPet. This illustrates how the potential for the Aequorea-based fluorescent proteins to self-associate can be an important consideration for many different types of studies using these cellular markers.
Conclusions
It took over thirty years, and the advent of recombinant DNA as well as vastly improved molecular biological approaches to see the pioneering work of Osamu Shimomura developed into a useful tool for live-cell imaging by Doug Prasher and Martin Chalfie. Just in the past decade, however, we have witnessed a truly remarkable expansion in the palette of Aequorea-based fluorescent proteins, largely driven by the innovative studies from Roger Tsien's laboratory. We now have jellyfish proteins that span an 80-nanometer portion visible spectrum from deep blue to yellow-green, providing a wide choice of genetically encoded markers for studies in cell biology. Most of the fluorescent proteins that are commonly used today have been modified through mutagenesis to optimize their expression in biological systems. Continued efforts using directed evolution approaches will no doubt improve the spectral characteristics, photostability, maturation time, brightness, acid resistance, and utility of the fluorescent protein tags for cellular imaging.
Contributing Authors
Richard N. Day - Department of Cellular and Integrative Physiology, Indiana University School of Medicine, 635 Barnhill Dr., Indianapolis, Indiana, 46202.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.