







Introduction
The noted German physicist and instrument designer Ernst Abbe realized in 1873 that the resolution of optical imaging in microscopy was fundamentally limited by the diffraction of light as spherical wavefronts pass through the circular apertures of objectives and conjugate focal planes. Abbe's research had revealed that the ultimate resolution of a microscope was not constrained by the quality of the lenses; rather it was restricted by the wavelength of light passing through the instrument as well as the aperture angle of the objective. In formalizing the relationship, Abbe noted that a microscope could not resolve two objects positioned closer than λ/2NA, where λ is the imaging wavelength and NA (numerical aperture) is defined by the objective aperture angle and refractive index of the imaging medium. This hallmark equation defines the lateral resolution in the x-y plane, which is perpendicular to the optical axis of the microscope. Resolution in the axial dimension is at least twice as poor. The diffraction barrier hindered the performance of optical microscopes for several centuries and was considered a physical limitation that could not be overcome with the use of standard glass-based objectives. In recent years, however, several new approaches have emerged that circumvent the limits of diffraction in optical microscopes based on the reversible photoswitching of fluorescent probes to achieve what is now commonly referred to as superresolution imaging. Thus, a number of exciting and creative new techniques have been advanced that apparently have no fundamental limit in achieving high spatial resolution, therefore rendering it possible to resolve specimen features in terms of just a few nanometers.
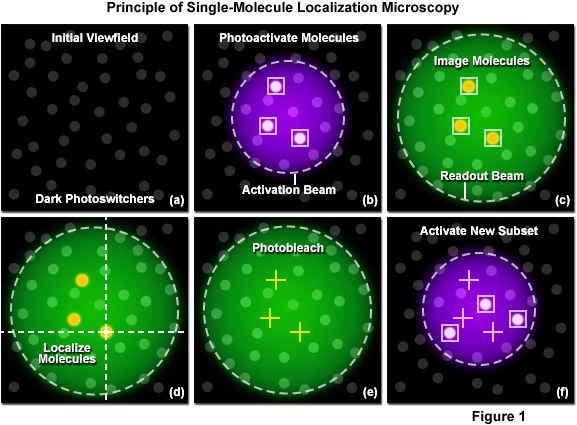
The new superresolution techniques can be roughly divided into two categories: those that are able to image ensembles of fluorophores using spatially modulated, focused light with saturating intensities, and those that sequentially image single molecule emitters that are spread to distances greater than the Abbe diffraction limit. The latter methods include photoactivated localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), and fluorescence photoactivation localization microscopy (FPALM), technologies that are differentiated primarily by the character of probes used for labeling the specimen. However, regardless of the minor technical differences between PALM, STORM, and FPALM, these single-molecule localization techniques all rely on the common principle of stochastically activating, localizing, and then photobleaching synthetic or genetically-encoded photoswitchable fluorophores. PALM and FPALM originally used photoactivatable or photoconvertible fluorescent proteins targeted to sub-cellular structures at high density to achieve the localization precision necessary to generate images at more than 10 times the spatial resolution afforded by diffraction-limited fluorescence microscopy. In contrast, STORM was developed using synthetic photoswitchable carbocyanine dyes to produce similar resolution.
The principle of single-molecule localization microscopy is illustrated in Figure 1 with a series of cartoons detailing the sequence of events for determining the precise location of a single set of photoactivated fluorescent probes. Initially all molecules in the specimen are inactive (native non-emissive state; dark circles) as shown in Figure 1(a). A violet laser is used to photoactivate a sparse subset of molecules, while a green laser is used for readout of the resulting fluorescent molecules. In Figure 1(b), a 405-nanometer laser is used to photoactivate a small number of molecules in the specimen (those surrounded by boxes). This number is maintained at a very low level by ensuring the laser intensity is sufficiently weak at the focal plane. Photoactivation of the molecules occurs stochastically where the probability of activation is proportional to location and the intensity of the activation laser. After photoactivation, the 561-nanometer readout laser is used to detect and record the position of the photoactivated molecules within the illuminated area (Figure 1(c)). Digital images of the photoactivated molecules are then analyzed to identify and localize molecules (Figure 1(d)) for as long as they remain fluorescent. During readout, the photoactivated molecules spontaneously photobleach (Figure 1(e)), eventually reducing the number of active molecules in the specimen. A new set of molecules is photoactivated (Figure 1(f)) to repeat the sequence, which is reiterated until all molecules in the specimen have been exhausted.
A number of practical considerations are mission-critical in achieving the best single-molecule localization images using PALM and related methodology. The primary concern is choice of photoactivatable probe, which will govern the maximum achievable resolution. This topic will be addressed in detail in the following sections. Second, although PALM instrumentation is relatively straightforward, some additional knowledge in the basic requirements for single-molecule imaging and other aspects of PALM are necessary to achieve successful results. Among the other important variables to understand for PALM imaging are the basic detector characteristics (pixel size, noise levels, intermediate magnification, readout speed), ensuring that a sufficient number of pixels are used to image single molecules, and maintaining the density of labeled features in the specimen at a level high enough to reconstruct the features of interest. Finally, careful attention must be paid to the details of specimen preparation to reduce autofluorescence and minimize potential artifacts induced by aberrations resulting from inhomogeneities and immersion media.
back to top ^Strategic Overview of PALM Imaging
PALM (as well as STORM and FPALM) is a single-molecule widefield technique where the raw data consist of an image stack typically containing thousands of individual frames, each featuring a subset of diffraction-limited fluorescence images of single photoswitchable molecules present in the specimen. The images of single molecules, appearing as bright point sources of varying intensity approximately 200 to 250 nanometers in width, are analyzed to determine their centers with high precision and the resulting information is employed to generate a high-resolution PALM image of the individual molecular coordinates. The resolution in PALM is thus dependent on localization precision, or how accurately the position of each single molecule can be determined from its diffraction-limited image. Perhaps more subtle, but equally important in determining the ultimate PALM resolution, is the molecular density of fluorescent probes in the specimen and how well they represent the target structure being analyzed.
The localization precision depends on achieving the maximum signal-to-noise ratio in each image, a characteristic that depends on maximizing the number of photons collected from each photoswitchable fluorescent probe while simultaneously minimizing the background fluorescence. Localization precision also depends on the total number of photons emitted by each molecule before it photobleaches, which is an intrinsic property of any particular molecule. In general, fluorescent proteins generate far fewer photons than do synthetic dyes, such as the carbocyanines (Cy3 and Cy5) or the ATTO dyes. In addition, the ability to take full advantage of the molecular density in each specimen is contingent on the contrast ratio of the probe, another intrinsic property that gauges the difference in fluorescence intensity between the on and off (or native and photoconverted) states of photoswitchable molecules. In many cases, the contrast ratio suffers from the fact that molecules supposedly in the "off" state often emit weak signals and contribute to the background noise. Therefore, the single most important factor to consider in designing an experiment using PALM imaging is the choice of fluorescent probe.
Although PALM and STORM were originally implemented using total internal reflection fluorescence microscopy, FPALM was originally reported using a widefield microscope that does not limit the focal plane to the proximity of the coverslip. However, all three techniques are compatible with both imaging modes and by applying typical widefield geometry, three-dimensional specimens can be imaged within a single focal plane of thickness approximately equal to the depth of field. This approach will work for sectioned tissues, bacteria, yeast, and other thin specimens, but suffers from significant background noise when attempting to image relatively thick tissue specimens using these single-molecule techniques where three-dimensional superresolution approaches are perhaps the best choice. PALM, STORM, and FPALM can be employed to gather a significant amount of single-molecule information about a specimen, including the number of photons emitted, intermittency, brightness, and the absolute number of fluorescent molecules. Additionally, the techniques can in principle determine emission spectra and fluorescence anisotropies of localized molecules.
In stark contrast to normal fluorescence imaging where specimens are initially very brightly fluorescent upon illumination, the single-molecule superresolution techniques are designed with probes that are in a dark or native emissive state (that is not imaged) before photoactivation. Therefore, virtually no fluorescence is emitted from a majority of the probes in PALM or STORM prior to being photoactivated or photoconverted by the activation laser. Once activated, the fluorescent probes are imaged with a longer wavelength readout laser (see Figure 1) matching their excitation properties. By carefully regulating the activation laser intensity, the rate at which molecules are activated can be controlled. One of the keys to PALM, STORM, and FPALM is the ability to turn off or disable the active molecules to avoid the number of emitters from growing so high that individual molecules are no longer distinguishable from one another. Removing active molecules from the field can be implemented through photobleaching or photoswitching to provide the critical balancing factor that limits the total number of visible molecules. In either case, this mechanism to control the number of active emitters is necessary to ensure that the distance between each active molecule and its nearest neighbor exceeds the lateral diffraction limit of 200 to 250 nanometers.
Once the density of fluorescent probes in a specimen is controlled to the point of allowing single-molecule imaging and localization, the only remaining requirement is a highly sensitive camera to capture images of the bright single emitters as they are activated and photobleached (or photoswitched to the off state). Photobleaching typically occurs spontaneously in the presence of the readout laser, but in cases where photoswitching is necessary, a second deactivation laser may be necessary. By creating a time-lapse video containing repeated cycles of photoactivation, readout, and photobleaching, the positions of a large number of molecules (ranging from several thousand to millions) can be determined. The uncertainty in the position of each molecule can also be determined by repetitive imaging of that molecule after it has been activated and before photobleaching. The resulting PALM image contains the measured positions of all the localized molecules displayed together.
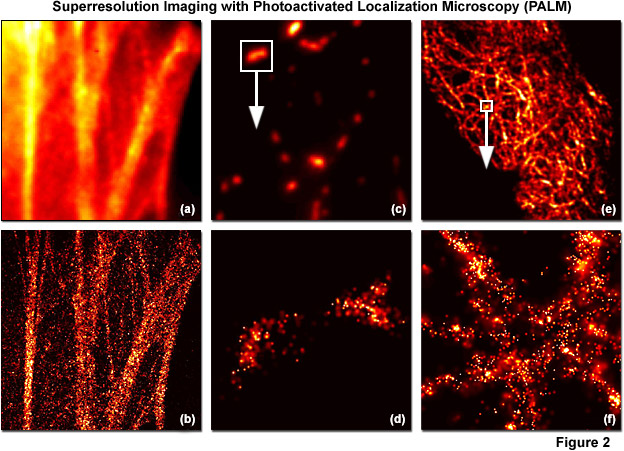
PALM images can be rendered either by plotting the unweighted positions of localized molecules or by using weighted plots of the positions of the localized molecules rendered as spots having Gaussian profiles where the intensity is proportional to the number of photons detected (from each molecule) and the radius is equal to the calculated or experimentally determined localization-based resolution. Because the weighted plots take into account the intensity and positional uncertainty of each molecule, the resulting images appear to the observer as a more realistic representation of a fluorescence image with high resolution (see Figure 2). Typically, all molecules localized within a specific area are rendered simultaneously, but in live-cell imaging or similar time-lapse sequences, time-dependent images can be created using subsets of molecules localized during various time periods. A threshold that includes only molecules featuring a particular range of intensities, or above a minimum intensity, can also be applied to increase the localization precision. In contrast, plotting only the unweighted positions of localized molecules leads to a plot of markers on a static background and appears more like a blueprint.
Illustrated in Figure 2 are summed widefield total internal reflection microscopy (TIRFM) and PALM single-molecule images of optical highlighter fluorescent proteins fused to targeting peptides or proteins that were localized within fixed mammalian cells. The filamentous actin cytoskeletal network is presented in Figures 2(a) and 2(b) using a fusion of tandem dimer Eos fluorescent protein (a green-to-red photoconverter) with human beta-actin and expressed in Gray Fox lung fibroblast cells. Note the individual fibers that are clearly resolved in Figure 2(b). In Figures 2(c) and 2(d), a single mitochondrion (outlined with the white box in Figure 2(b)) labeled with dimeric Eos fused to the mitochondrial targeting sequence from subunit VIII of human cytochrome C oxidase is shown in widefield (Figure 2(c)) and PALM (Figure 2(d)). Finally, in Figures 2(e) and 2(f), the intermediate filament network in HeLa cells is highlighted with human cytokeratin fused to mEos2, a monomeric version of Eos fluorescent protein. The boxed area in Figure 2(e) is shown in PALM to demonstrate molecular locations in Figure 2(f). In all cases, photoconversion was conducted with a 405-nanometer diode laser and readout occurred using a 561-nanometer diode-pumped solid state laser.
back to top ^Localization Precision
In widefield and confocal fluorescence microscopy, whenever two molecules are confined within the same diffraction-limited diameter of the point-spread function (approximately 250 nanometers), they cannot be distinguished as separate individual emitters. The resulting images are blurred by diffraction with a loss of any features smaller than the point-spread function. On the other hand, it has been well established that single fluorescent molecules can be localized with a high degree of precision (much smaller than the diffraction limit) as discussed above. Localization of each molecule essentially amounts to a summed measurement of its lateral position, where each photon that can be detected from any specific molecule constitutes a single measurement of that position. Thus, as more photons are detected, the result will be increasingly better localization precision. The concept behind localization precision can be quantitatively expressed in Equation (1):
σ2x,y = [(s2 + q2/12) / N] + [(8πs4b2) / (q2N2)](1) σ2x,y ≈ s2 / N(2)
where σx,y is the localization precision of a fluorescent probe in the lateral dimensions, s is the standard deviation of the point-spread function, N is the total number of photons gathered (note: this does not represent the number of photons per pixel), q is the pixel size in the image space, and b is the background noise per pixel (not the background intensity). When conducting PALM experiments with small pixel sizes (similar to those found in high-performance cameras) and negligible background noise from dark emitters and autofluorescence, Equation (1) can be approximated by Equation (2), which can be considered in this situation as governing the localization precision of single molecules imaged by fluorescence microscopy.
Several important guidelines should be followed for optimizing the localization precision (in effect, minimizing σx,y). Perhaps the most important consideration is to maximize the number of photons gathered (N) because both terms in Equation (1) decrease as N increases. The first term on the right-hand side of Equation (1) accounts for the contribution to the localization uncertainty from shot noise and pixelation noise in the absence of background signal. However, even when there is no confounding background, a molecule imaged with N detected photons will appear noisy and spread with a width approaching the diffraction limit. In order to localize the centroid of the diffraction-limited spot, the image is typically analyzed by applying a least-squares fit to a Gaussian approximation of the point-spread function (or the function itself if that can be determined). Presented in Figure 3 are simplified illustrations of the single-molecule localization process. The image of a single emitter (considered raw data; Figure 3(a)) is mathematically treated to fit a two-dimensional Gaussian function and localized with nanometer accuracy (Figure 3(b)). The entire process is schematically depicted in Figure 3(c) to show the Gaussian profile of a point-spread function sandwiched between the raw and processed data.
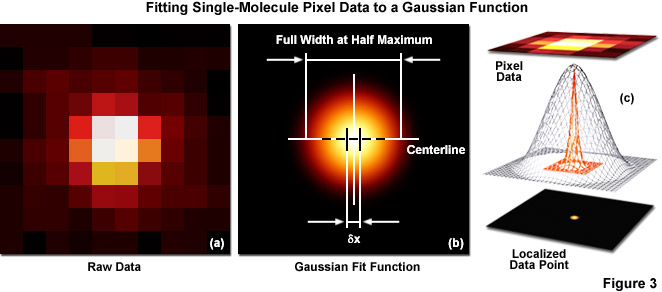
Increased noise levels will result in more uncertainty about the position of a single molecule, but the localization precision will increase as the number of detected photons increases (thus, as discussed above, probe choice is of paramount importance). The pixel size (q) also exerts a small influence on the equation such that for larger pixel sizes, the uncertainty in localization precision increases. In the extreme case of using pixels much larger than the diffraction limit due to a mismatch of camera and microscope optical resolution (the unusual case of having pixels greater than 250 nanometers in width), the size of the pixel itself governs the localization uncertainty because the exact position of the molecule within the pixel is not clear. In practice, however, the pixel size becomes negligible when q becomes much smaller than s, but due to readout noise and other factors, the value for q should not decrease dramatically below the value for s. Thus, the investigator should ensure that intermediate magnification is installed in the microscope optical train to ensure the pixel size conforms to the Nyquist limit.
The second term on the right-hand side of Equation (1) encompasses the localization uncertainty produced by background noise resulting from a number of sources including camera electronics, autofluorescence, scattered light, non-specific labeling, and weak fluorescence from inactive probe molecules (supposedly switched off or not yet photoconverted), as well as a variety of other sources. Light detected from sources other than the molecule of interest will lead to a higher level of noise in the image and degrades localization precision. The second term in Equation (1) therefore accounts for the background noise per pixel (b) in photons rather than the background signal level. A truly uniform offset value can be subtracted and will not add to localization uncertainty. This type of offset is often referred to as the black level setting in a digital camera and is applied to all pixels in the image. In summary, minimizing the background noise while simultaneously ensuring the greater number of detected photons is the key to successful results in PALM imaging. Note that the second term in Equation (1) depends on the detected photons as the reciprocal squared, so the relative improvement as N increases is more significant for the second term compared to the first term.
In order to reduce the effect of background noise in PALM imaging (and increase the localization precision), sources of unwanted photons should be eliminated or, at the very least, attenuated to the largest degree possible. Prior to data acquisition, the image beam pathway should be shielded to minimize external background light from reaching the camera image sensor. Common internal (specimen and microscope) sources of background noise include oil or water immersion media, objective front lens contamination, autofluorescence from the coverslip, tissue culture medium, or transfection reagents, fluorescence from inactive photoswitchable probes, emission from active fluorophores that are removed from the focal plane (this effect is minimized when imaging in total internal reflection mode), and scattered laser light. Dark and read noise from the camera can also be a significant contributor to the background, although these effects are usually negligible in high-performance electron-multiplying digital cameras operated at high gain. The presence of cellular autofluorescence can result in a significant position-dependent background signal that decreases over time due to photobleaching. Background can also be efficiently reduced by careful choice of filters for the image beam to exclude wavelengths outside the emission passband.
back to top ^Molecular Density
A quite useful feature associated with single-molecule localization techniques is that the investigator can define which molecules are displayed by choosing a lower limit on the localization precision. Thus, the investigator may decide to display only molecules that localized with better than 10-nanometer accuracy and discard those with inferior precision. In this case, the difference between localization precision and the resolution obtainable with single-molecule localization must be explained. Aside from localization precision, the other key determinant of resolution in PALM imaging is the density of labeled molecules in the specimen. According to the Nyquist-Shannon sampling theorem, the sampling interval (mean distance between neighboring localized molecules) must be at least twice as fine as the desired resolution, or two data points per resolution unit. Otherwise, the feature of interest will be undersampled and not resolved. As an example, to achieve 10-nanometer lateral resolution, molecules must be spaced a minimum of 5 nanometers apart in each dimension to yield a minimum density of 40,000 molecules per square micrometer (or approximately 2,000 molecules in a diffraction-limited region of 250-nanometers in diameter).
Presented in Figure 4 is the concept of molecular density in superresolution imaging to demonstrate the fact that in addition to nanometer localization precision, a high density of localized molecules is necessary to reveal many of the essential structural details. The specimen in Figure 4 consists of a series of three simulated nanoscale structures having different labeling densities that lead to PALM images having varying levels of resolved molecular density. The pattern in each simulation is a line structure with stripes of 1-micrometer width and increasing density as observed from Figure 4(a) to Figure 4(c). In each pattern, the red dash outline in the upper box corresponds to the zoomed region shown in the lower histogram graph that plots the number of localized molecules versus distance. Note that the red outline starts in a dark region devoid of molecules and spans into the center of the adjoining stripe. The simulated patterns contain 1694, 16732, or 148790 molecules (Figures 4(a), 4(b), and 4(c), respectively) localized in a background-free area approximately 8 square micrometers in size. The histograms illustrate the number of molecules localized within evenly spaced bins to reveal the lateral profile at the edges of the stripes. When only a limited number of molecules are localized (Figure 4(a)), the ability to discern the edges is limited, whereas at very high molecular densities (Figure 4(c)), the lateral profile of the edge is clearly visible, even though the localization precision was the same in all three cases.
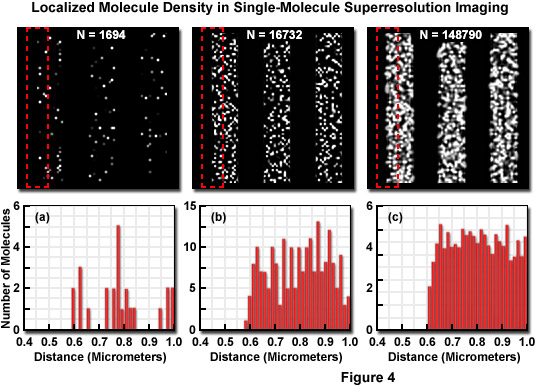
In many circumstances, superresolution experiments are limited based on the specimen and the fluorescent molecule of interest rather than an arbitrarily imposed limitation on molecular precision. This can occur from a normal low density of molecules within the specimen due to the simple fact that scant targets exist for primary antibodies when using immunofluorescence or the targeting protein in fluorescent protein fusions has a low abundance level. Similar to the argument presented above, for a photoactivated fluorescent protein with a diameter of approximately 2.5 nanometers in a two-dimensional plane (such as an organelle membrane), the maximum density of molecules is around 200,000 per square micrometer. In order to reach such a high density, it must be assumed that only fluorescent protein fusion molecules are located within this plane, which is unrealistic. Molecules constrained to polymers may approach this level, but such a high concentration of any single molecule is an unlikely physiological situation.
Alternatively, if fluorescent protein fusions comprise 1 percent of the molecules in a membrane (a more reasonable concentration), this leads to approximately 2,000 molecules per square micrometer or around 2 molecules every 40 to 50 nanometers. This relatively low concentration limits any resolution claims about structural features. Nevertheless, experiments observing fluorescent probes at this concentration in an organelle membrane offer a significant amount of information, despite the limits on resolution. For example, they can reveal relative molecular distribution within the specimen, which is often the goal of an imaging experiment. As PALM techniques are extended to dual-color imaging, overlaying images of two different proteins localized in the same specimen will ultimately yield far more useful information at low molecular densities.
back to top ^Fluorescent Probe Choices for PALM
Single-molecule localization superresolution imaging can be conducted using a wide variety of fluorophores, including fluorescent proteins, synthetic dyes, caged compounds, quantum dots, and hybrid systems that couple a genetically-encoded target peptide to a membrane-permeant neutral fluorescent ligand that activates upon reaction with functional groups in the target. Fluorescent proteins have the distinct advantage of being exquisitely specific in their targeting. In effect, by genetically fusing a photoswitchable fluorescent protein to a peptide, signal sequence, or protein of choice, the investigator is assured that there exists a one-to-one correspondence between a detected molecule and its target, thus avoiding nonspecific binding that often arises with antibodies or proteins conjugated to synthetic dyes. Furthermore, targeting is in general far more accurate with fluorescent proteins than with many reagents used to fluorescently label cells and tissues, and usually exhibits lower levels of background fluorescence at low to moderate expression levels. Synthetic fluorophores and quantum dots conjugated to antibodies will not pass through the plasma membrane and can only be used to highlight internal cellular structures after treatment of the cells with a detergent, which will often induce additional artifacts including autofluorescence.
Fluorescent proteins better approximate the size of a target protein, whereas antibodies can introduce considerable uncertainty with respect to localization coordinates due to their size (ranging from 10 to 20 nanometers, depending on whether both primary and secondary antibodies or fragments are used in labeling). The probe density should be made as high as possible within the constraints of standard fluorescent specimen preparation. One of the primary considerations is that overstaining with synthetic fluorophores (and using immunofluorescence) or excessively high expression levels with fluorescent proteins should be avoided in order to prevent disruption of biological structures of interest and to maintain low background fluorescence levels. In addition, the probe density should be low enough to image distinct molecules with a large enough separation distance for precise localization. If the fluorescent probe exhibits spontaneous activation with the readout laser, there will be emitting molecules even before deliberate photoactivation has been initiated. In many cases, the specimen can be irradiated to intentionally photobleach a subpopulation of the fluorophores to reduce background levels.
Using the large cadre of currently available photoactivatable or optical highlighter fluorescent proteins, cells are typically transfected with a construct containing the DNA encoding the fluorescent protein fused to the gene for the protein of interest. Probes can be specifically tailored for individual biological applications by adjusting the promoter strength or using alternative molecular biology techniques, such as a viral host or newly introduced point mutations to improve performance. Such alterations can modulate the fluorescent protein expression level, the absorption and emission spectra of the highlighter, sensitivity to pH or ion concentration, various environmental parameters, or a host of other useful properties. Thus, given the flexibility in working with genetically encoded markers, the investigator can control many of the experimental variables to a large degree. The situation is less favorable with synthetic dyes and quantum dots, where targeting is an issue and fluorescence intensity levels are dictated by the concentration of labeled secondary antibody or the dissolved dye. In this case, the optimum labeling density can readily be determined by trial and error.
A number of criteria are desirable for photoactivatable or photoswitchable probes used in single-molecule localization, regardless of the nature of the particular fluorophore. The ideal probes should have large extinction coefficients and quantum yields at the activation and readout wavelength. These two quantities are multiplied to determine the brightness of the probe, with the highest brightness levels being the most advantageous. There should also be a reduced tendency for self-aggregation, manifested by dimerization or oligomerization in both synthetic dyes and fluorescent proteins. Aggregation is much less of a problem in specimens prepared using immunofluorescence techniques. When recording fast events with independently running acquisition in single-molecule imaging (known as PALMIRA), a reasonable quantum yield for readout-induced activation is necessary. In terms of photobleaching, the rate should be small, but finite, because the active probes must emit a large number of photons yet still be able to eventually photobleach to avoid the density becoming too high to allow individual identification and localization.
Regardless of the labeling strategy or the probes involved, one of the most important factors that influences both the maximum achievable density and localization precision is known as the contrast ratio of the photoswitchable fluorescent label. This quantity is defined as the ratio in fluorescence intensity between the inactive and activated states of the probe. If the contrast ratio is less than the density of the probe in any particular diffraction-limited region, the weak intensity from the inactive molecules can overwhelm the intensity from a single activated molecule, complicating both localization and measurement of densely labeled specimens. The contrast ratio has not been measured for many of the currently available probes reported to be useful in single-molecule superresolution imaging, but the carbocyanine dyes have been determined and can achieve very large ratios. In addition, the tandem dimer version of Eos fluorescent protein (tdEos) has a higher contrast ratio than many of the other fluorescent proteins used in PALM imaging. A systematic investigation of photoswitchable probe contrast ratios would prove very beneficial to the scientific community.
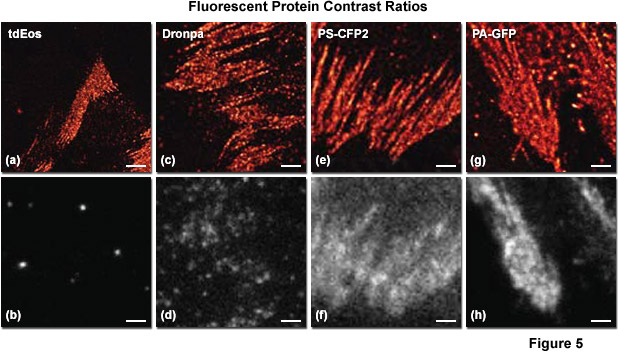
Presented in Figure 5 is a comparison of contrast ratios for several optical highlighter fluorescent proteins used in PALM imaging. The specimens were all labeled with the appropriate fluorescent protein fused to paxillin, which localized the fusion to focal adhesions. Images in the upper row feature PALM results in each case, whereas the lower row shows frame number 1000 obtained from the stack of single-molecule images used to generate the corresponding PALM image. Thus, for example, tdEos has the highest contrast ratio (Figures 5(a) and 5(b)), whereas PS-CFP2 (photoswitchable cyan fluorescent protein, version 2; Figures 5(e) and 5(f)) and PA-GFP (photoactivatable green fluorescent protein; Figures 5(g) and 5(h)) have the lowest. In all cases, background fluorescence signal largely reflects emission from the large pool of inactive (native) fluorescent proteins. Note the correlation between low background (in effect, high contrast ratio between the active and inactive states) and the sharpness of the resulting PALM image, which is a measure of localization precision.
In review, each class of fluorescent probes has its particular strengths and weaknesses, however no single class or individual fluorophore has yet been developed that combines all the preferred characteristics of an ideal probe for any application in single-molecule superresolution microscopy. The fundamental aspect for choosing superresolution probes is that they must be capable of being either photoactivated, photoswitched, or photoconverted by light of a defined wavelength band as a means to alter their spectral properties for the detection of selected subpopulations. Besides displaying the necessary fluorescence emission and other photophysical properties, PALM superresolution probes must also be capable of localizing to their intended targets with high precision and exhibit the lowest possible background noise levels. Fluorescent proteins, hybrid systems, and highly specific synthetic fluorophores (such as MitoTrackers) are able to selectively target protein assemblies or organelles, but most of the cadre of synthetic dyes and quantum dots must first be conjugated to a carrier molecule for precise labeling.
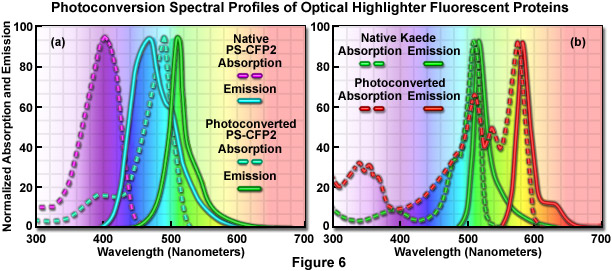
The absorption and fluorescence emission spectral profiles of two optical highlighter fluorescent proteins used in PALM imaging are shown in Figure 6. The native and photoconverted states of the fluorescent protein, PS-CFP2, are drawn in Figure 6(a). The absorption spectra (maxima at 402 and 490 nanometers, respectively) are indicated by violet and cyan dashed lines, while the emission spectra (respective maxima at 468 and 511 nanometers) are noted with cyan and green solid lines. There is sufficient separation of the cyan and green spectral profiles to use this probe in single-molecule superresolution imaging. Likewise, the absorption and fluorescence emission spectral profiles of natural and photoconverted Kaede fluorescent protein are presented in Figure 6(b). The non-converted Kaede absorption and emission spectral profiles are outlined in the graph with green dashes and lines, respectively, while the photoconverted Kaede profiles are denoted with red dashes and lines. Note the relatively small Stokes shift (approximately 10 nanometers) for both pairs of spectra, but the large separation distance (60 nanometers) between the green and red emission maxima.
In addition to having high brightness levels, the best probes for superresolution microscopy should exhibit spectral profiles for the active and inactive species that are sufficiently well separated (see Figure 6) and thermally stable so that spontaneous interconversion energies are very low compared with the light-controlled activation energy. Ideally, these probes should also exhibit high switching reliability, low fatigue rates (in effect, the number of survivable switching cycles), and switching kinetics that can be readily controlled. In terms of photobleaching or photoswitching to a dark state, the best probes are those whose inactivation can be balanced with the activation rate to ensure that only a small population of molecules is activated (to be fluorescent) for readout, and that these activated molecules are separated by a distance greater than the resolution limits of the camera system. Furthermore, each photoactivated molecule should emit enough photons while in an activated state to accurately determine their lateral position coordinates. Provided that these generalized guidelines are followed, the investigator should have success in PALM imaging.
back to top ^Instrument Considerations
Single-molecule superresolution microscopy techniques, including PALM and STORM, require efforts to suppress background fluorescence in order to optimally detect the relatively faint emission from individual probes. One of the most useful microscopy techniques for this application is total internal reflection fluorescence (TIRF) microscopy because the evanescent excitation wave penetrates only approximately 200 nanometers into the specimen (about the same width as the point-spread function), leading to extreme rejection of the background fluorescence common in most applications. TIRF is also a widefield technique, implying that many molecules can be imaged simultaneously in each frame, thus vastly increasing the acquisition speed. Additionally, TIRF is an ideal candidate for imaging fluorescently labeled structures that lie close to the membrane, such as focal adhesions, endosomes, clathrin, and cytoskeletal components. Note that TIRF is not an absolute requirement for PALM or STORM imaging as certain specimens (such as bacteria) are sufficiently thin that autofluorescence does not impede the localization of single molecules illuminated in epifluorescence.
Many of the existing commercial TIRF microscope configurations can be utilized for PALM imaging after some minor modifications and additions to the existing hardware, but new turnkey instruments designed specifically for superresolution imaging have been introduced and will be easier to configure for many investigators. Specific hardware components requiring specialized attention for superresolution imaging include the laser systems, objectives, a high-resolution mechanical stage, vibration isolation, detectors, filters, and the data acquisition computer. Free-space coupling (without an optical fiber) of appropriate excitation and activation lasers into the microscope ensures that maximum power is delivered to the specimen to permit the fastest possible imaging. Alternatively, fiber-coupled lasers having power outputs of 50 milliwatts or more can be employed. Objectives should be chosen with high numerical apertures to enable the best possible photon collection efficiency, and the appropriate camera system (discussed below) should be employed. Excitation, emission, and dichromatic interference filters should be of the highest quality with passbands tuned to the photophysical characteristics of the probes in use. Also, as is the case in all forms of single-molecule microscopy, the instrument should be perfectly aligned with the optical surfaces meticulously cleaned. Finally, a high-performance computer with acquisition software, image capture board, and a large hard drive is critical for acquiring and storing the often exceedingly large PALM datasets.

Illustrated in Figure 7 is a state-of-the-art inverted microscope designed for imaging in laser scanning confocal and single-molecule PALM modes. The ZEISS Axio Observer frame is equipped with a dual-camera adapter that couples two Andor iXon 897 electron-multiplying CCD cameras for imaging in two channels. A laser scanning module, the LSM 780, allows the specimen to be probed with point-scanning confocal microscopy in thin sections. The high-performance scanning stage features a piezo insert that enables precise control of the specimen location in the axial dimension. The ELYRA module contains turnkey components that allow the operator to image a selected region of the specimen with TIRF to gather PALM images for superresolution analysis. Separate laser systems for the confocal and PALM units afford flexibility with regards to the excitation lines necessary to conduct both imaging techniques. Finally, the microscope is equipped with software that enables image assembly and analysis with both confocal and PALM imaging modalities.
The choice of objective will often dictate other instrument parameters. For TIRF imaging in PALM and STORM, the numerical aperture of the objective must be greater than the refractive index of the medium bathing the specimen (usually a dilute aqueous buffer with a value approximately 1.33 to 1.38). In practice, number of objectives that satisfy this criterion are limited to those having a numerical aperture greater than 1.45 and specifically designed for TIRF imaging. High numerical aperture objectives typically exhibit very good photon collection efficiency and also feature an increased range of angles over which total internal reflection can be readily achieved. TIRF objectives usually have a wider annulus in order to provide a large adjustment range of the evanescent wavefield decay characteristics, making it easier to achieve TIRF by focusing a spot at the objective rear focal plane without clipping the beam. The choice of immersion oil and coverslip thickness is also important. Most high numerical aperture objectives are designed for use with 170-micrometer (number 1.5) coverslips, and index-matched immersion oils can be obtained from commercial manufacturers. Each immersion oil should be carefully checked for autofluorescence or production of aberrations in the point-spread function.
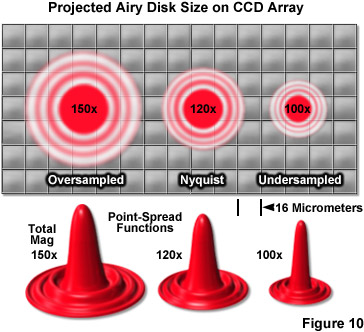
Objective magnification should be selected to match the pixel size of the detector when coupled with the appropriate tube or relay lens, as described below. The final configuration should produce images of single molecules on the detector array that span at least two pixels, but not substantially more in order to avoid readout and background noise from interfering with localization calculations (see Figure 10). For example, for a digital camera having 16-micrometer pixels, the effective pixel size at the specimen plane is approximately 270 nanometers and can be roughly determined by dividing the camera pixel size by the objective magnification. When dealing with large effective pixel sizes, localizing individual molecules becomes more difficult because of the uncertainty of the exact location of the molecule within the range of a large pixel. Essentially, when the image of a single molecule does not span more than a single pixel, it is not possible to determine where the image is located more accurately than within the dimensions of that pixel. In the ideal case, the image should span approximately 9 pixels (a central pixel with 8 neighbors involved) so that the relative level of intensity in each of the adjacent pixels can be used to more accurately determine the molecular position. Thus, the basis for determining the molecular localization with a precision exceeding one pixel is the ability to use intensity from adjacent pixels in the calculation.
PALM and STORM microscope configurations require specialized interference filters and dichromatic mirrors of the highest quality to maximize fluorescence signal while rejecting unwanted excitation light to ensure the best signal-to-noise ratios and eliminate or reduce stray light and autofluorescence. Because lasers are used for photoactivation and readout (see Figure 8), laser cleanup filters having a narrow bandwidth (in the range of 10 nanometers) are often useful. Dual or triple band dichromatic mirrors are employed to simultaneously reflect both activation and excitation wavelengths in order to avoid wasting time switching single band mirrors during the experiment. Dichromatic mirrors should also be capable of transmitting the much weaker fluorescence emission with high efficiency. Emission filters must provide at least six optical density units of excitation light suppression in the out-of-band regions while maximally transmitting the fluorescence emission. Bandpass filters are usually the best choice (as opposed to longpass or shortpass filters) and high-quality filters of varying designs are readily available from the manufacturers. When selecting filters, the new hard coat filters offer steeper cut-on and cut-off slopes, as well as higher transmission. They are marketed under a variety of special brands and are produced by all of the major filter manufacturers.
For excitation and photoactivation or photoconversion, PALM and STORM instruments should be equipped with solid state diode lasers having output power ranging between 50 and 200 milliwatts to provide sufficient energy for single-molecule photoswitching and imaging. Most of the fluorescent proteins and several of the synthetic dyes used in these superresolution techniques are efficiently activated at 405 nanometers, so a laser in this wavelength range is quite useful. Other laser wavelengths that match the typical fluorophores for single-molecule imaging have emission lines at 488, 561, 594, and 635 nanometers. Powerful gas lasers can also be used, but these sources are considerably more expensive, feature shorter lifetimes, and often require water or forced air cooling. A note of caution surrounding diode lasers is that the beam mode quality may be poor and the intensity may drift slightly (especially with some 405-nanometer lasers), but for PALM, these characteristics are not critical so long as enough power is available to excite or activate probes in the desired specimen region. However, the proper configuration for TIRF may prove difficult to achieve when attempting to co-align a poor beam profile with another laser.
For maximum power throughput, the lasers can be free-space coupled to the microscope illumination port (Figure 8), with neutral density filters and half-wave plates to control the intensity and polarization azimuth (the latter is important for maximum transmission through an acousto-optic tunable filter; AOTF). As mentioned above, newer high-performance laser combiners that have an average output power in the 50+ milliwatt range are expensive, but preferred. Lasers can be filtered through a narrow bandpass excitation filter to reduce emission noise. For multiple lasers, beam expanders placed after each laser can generate a common beam diameter. In commercial PALM systems, many of the considerations just described have been addressed in other ways by the manufacturers, who carefully engineer their instruments to produce single or multi-line laser outputs that are more conveniently adjusted to obtain the desired critical angle. Furthermore, turnkey PALM instruments are pre-equipped with filter sets and objectives designed to optimize the performance of these microscopes for single-molecule imaging at high frame rates in order to obtain the best possible image quality.
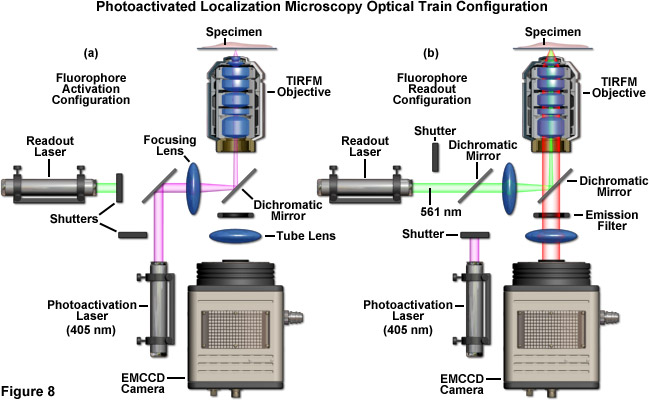
A schematic illustration of a typical PALM microscope configuration is presented in Figure 8. The activation laser is directed into the microscope using a dichromatic mirror and is positioned to be co-linear with the readout laser. Both beams are focused through the same lens and reflected by a second dichromatic mirror to form a focal point in the rear aperture of the objective, which causes a large region of the specimen to be illuminated. The emitted fluorescence is gathered by the single objective and is transmitted through the dichromatic mirror and an emission filter. Emission is focused by a tube lens to form an image of single molecules on the camera area-array detector. The activation and readout lasers are controlled by shutters (or optional AOTFs) and various mirrors are used for steering the light beams. For the greatest economy, neutral density filters (not illustrated and not recommended) can be used to attenuate laser power.
Microscope stability is a critical factor in all forms of single-molecule imaging, including PALM, STORM, and related techniques. Even slight lateral drift (measured in nanometers) can significantly interfere with the precise localization of single emitters and axial drift can completely wipe out signal if the specimen moves away from the focal plane. In general, a motorized microscope firmly mounted on an air-suspension vibration isolation platform on a solid concrete slab will provide reasonable positional stability over timescales of several minutes. A typical configuration will feature lateral drift of approximately 6 to 7 nanometers after a period of 20 minutes, so localization precision rather than drift will dominate the resolution of images in PALM using fluorescent proteins for relatively short periods (30 minutes or less). Longer acquisition periods will require the application of fiduciary markers (such as quantum dots, colloidal gold, or fluorescent microspheres) to compensate for stage drift. In addition, automatic focus drift correction hardware, which is offered by the major microscope manufacturers, is useful to maintain the specimen within a few nanometers of the axial focal plane. This feature is especially important for densely labeled features that will have fluorescent molecules scattered across multiple focal planes.
back to top ^Detector Configuration Parameters
Because PALM, FPALM, STORM, and related widefield superresolution microscopy techniques all involve single-molecule methodology with its inherently low signal levels, careful selection of a photon detector is of paramount importance. Unlike the case for laser scanning confocal or multiphoton microscopy, both of which successfully utilize photomultiplier point-source detectors, the widefield illumination patterns generated in single-molecule imaging mandate the use of an area-array imaging device, such as a scientific-grade digital camera. It is therefore critical to select the proper detector in order to achieve the best results in PALM imaging, but the number of commercially available detectors is enormous and the correct choice can often be confusing. The two most dominant technologies are charge-coupled device (CCD) and scientific-grade complementary metal oxide semiconductor (CMOS) based digital cameras.
High-performance CCD cameras offer superior quantum efficiency (number of photoelectrons generated per incident photon), usually in the range of 90 percent or higher across the visible spectrum, when compared to the CMOS cameras that usually have values peaking at approximately 15 to 25 percent in the same spectral region. On the flipside, the frame rates of CMOS cameras dramatically exceed those of CCD-based devices, such that the former are more useful when signal levels are relatively significant and high-speed imaging is necessary. Generally, slower scientific-grade CCD cameras are relegated to applications requiring low-signal time-integrated measurements. Therefore, the requirements of single-molecule imaging appear to lie squarely in the middle between the capabilities of these two technologies, requiring both high quantum efficiency and relatively fast frame rates.
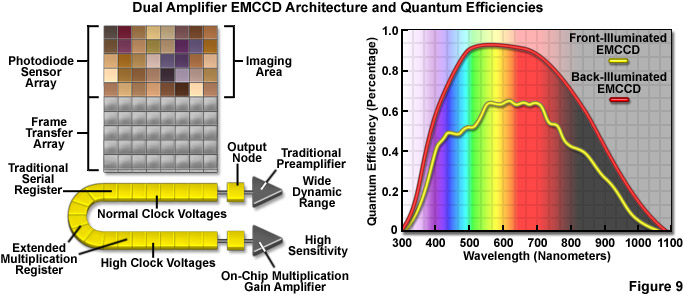
Recent design advances have improved the quantum efficiency as well as a number of other aspects in the performance of CMOS devices, and newer CCD cameras (even those with extremely high sensitivity) have dramatically improved frame rates. Some of the best CMOS cameras now sport quantum efficiencies in excess of 45 percent and CCD cameras are commercially available with frame rates up to 10,000 per second. However, at this point the most useful cameras for single-molecule imaging are electron-multiplying CCDs (EMCCDs) that offer both high frame rates (greater than 30 per second) and extremely low readout noise, with the ability to operate in frame-transfer mode (spooling acquired data to disk while simultaneously acquiring new data). Most EMCCD cameras can be obtained with sensors that are back-illuminated where incoming photons impact on the rear side of a mechanically thinned CCD instead of the front side for greater quantum efficiency (see Figure 9). The cameras are also offered with different pixel dimensions, array sizes, and readout speeds. Advanced EMCCD models offer dual readout registers that feature a traditional preamplifier for capturing images with wide dynamic range (Figure 9) as well as the on-chip multiplication gain amplifier for high sensitivity single-molecule applications.
EMCCDs perform by applying gain (amplification) during the last stage of the photoelectron line-shifting process, but prior to amplified electrons entering the analog-to-digital conversion circuitry. By amplifying the signal on the CCD chip before readout, the read noise associated with the amplification process in conventional CCDs is greatly reduced so that this noise source no longer limits sensitivity. EMCCD chips have a noise component, termed clocking induced charge (CIC), which has no equivalent in normal digital cameras. The phenomenon occurs when electrons are being transferred through the multiplication register under the influence of high-voltage clocking pulses, and is usually treated similarly to dark current. Most of the major scientific camera manufacturers offer high-performance EMCCD-based digital cameras, many of which are cooled to between -70° and -100° C to almost completely eliminate dark current. Before selecting a camera for PALM imaging, commercial EMCCDs should be carefully examined and tested under the prevailing experimental conditions.
Due to the fact that single-molecule imaging can be considered to be a seriously signal-starved application, the fluorescence emission rate and microscope photon collection efficiency are able to place an upper limit on the camera frame rate. Thus, in order to achieve a resolution that is an order of magnitude beneath the diffraction limit (approximately 20 nanometers), the shot noise statistics of photon counting require that a minimum number of photons be recorded for any particular molecule that is being localized. As an example, for 100 photons, this requirement corresponds to single-frame exposure times in the range of approximately 100 milliseconds, which lies well within the capabilities of an EMCCD camera. It is important to note, however, the difference between exposure time and frame rate when comparing camera specifications.
Expensive cameras with large numbers of pixels that are also equipped for electronic gating are often referred to as "fast" with regards to their ability to rapidly switch between active and inactive CCD states, rather than their ability to quickly obtain sequential images. In such cameras, the frame rate and the exposure time differ by what is termed the dead time necessary to clear the CCD chip of residual charge, transfer the image information, and prepare the chip for the next exposure. The dead time is a function of the number of pixels in the CCD array. For single-molecule PALM applications using fixed specimens, a frame rate around 20 per second or greater is most useful, whereas for live-cell imaging, frame rates exceeding 100 are often necessary.
Digital camera detection efficiency is another important aspect to consider when configuring a microscope for PALM experiments. As mentioned above, the intrinsic quantum efficiency of a detector is defined as the fraction of absorbed photons that are converted into an electrical signal. Such a simplistic definition accounts only for the electronic conversion efficiency and ignores other important properties, such as the efficiency of the photon absorption process, which is wavelength dependent. As a result, use of the term external or total quantum efficiency is often substituted to describe the combination of all signal losses and can be quantified using a broadband light source. The total quantum efficiency takes into consideration the fill factor of the CCD, which is the percentage of real estate on the chip that is able to absorb incoming photons. EMCCDs have the highest fill factor, followed by interline CCDs (found in most cooled scientific cameras) and CMOS-based cameras. For single-molecule imaging, cameras with the highest fill factor are necessary, especially for dim specimens. The highest quality back-thinned CCD devices have excellent quantum efficiencies (around 90 percent) in the central region of the visible spectrum, but this value falls at both extremes. It is therefore important to ensure that digital cameras selected for single-molecule imaging feature high fill factors and quantum efficiencies as high as possible in the spectral region corresponding to the emission frequencies associated with the fluorescent probe(s) of interest.
Digital cameras are associated with a variety of noise sources, several of which are very important for single-molecule localization as outlined in Equation (1). Some of these noise sources are associated with the CCD image sensor, while the others are characteristic of the camera electronics. To better understand the situation with regard to localizing individual molecules, consider imaging approximately 100 photons emitted by a single molecule that are captured by a CCD camera. If the photons are distributed over a 9-pixel area and the point-spread function is approximately Gaussian, then the number of photons incident on the central pixel can range from 30 to 50. The number of photons on the peripheral 8 pixels will average 6 to 10. For these peripheral pixels, fluctuations in the background signal level are expected to play a more significant role in localization (term b in Equation (1)). As will be described in the following paragraphs, the two most significant background noise sources associated with CCD cameras are dark current and read noise.
In CCD imaging sensors, dark current is generated by impurities and defects in the bulk semiconductor or at the gate interfaces. At sufficient thermal energy, electrons can migrate between the conduction and valence bands to increase the apparent electronic signal generated in a pixel, even in the absence of light. Because dark current is temperature sensitive, an effective solution to reduce this noise source is to cool the CCD with a thermoelectric (Peltier) device. Using Peltier-cooled CCDs at temperatures below approximately 40° C, the effective dark current can be lowered to 1 electron per pixel per second or less. In many imaging applications, dark current can be treated as a systematic background offset and subtracted by using a reference image acquired with no incoming signal. However, the noise associated with dark current cannot simply be subtracted with a reference and, similar to shot noise, is equal to the square root of the dark counts. As a result, in cameras having a large dark current, the noise can result in fluctuations of comparable magnitude to the single-molecule signal at each pixel, producing a serious problem. Therefore, the best solution for dark current is to use an EMCCD camera with external cooling to very low temperatures.
Another factor to consider with regards to dark current is the non-uniformity associated with each pixel due to manufacturing variations that result in variations in the density of defects and impurities from one pixel to another. Thus, each pixel will exhibit a slightly different voltage offset, sensitivity to light, and rate of dark current production. Such a variation between pixels results in fluctuating levels of dark noise, and is referred to as dark noise non-uniformity. As CCD arrays continue to increase in physical size and pixel dimensions simultaneously decrease, the relative variations in defect density are expected to become more significant and should be considered when purchasing new, large area-array CCDs. Dark noise non-uniformity imposes a variance in the background noise value (b) described in Equation (1), which results in an increase in localization uncertainty. The variations in average dark noise can be determined by sampling a large number of dark frames and calculating the average dark noise for each pixel, as well as the statistical variance across all pixels.
The process of extracting and converting the photoelectron charge at each pixel into an electrical signal followed by conversion of that voltage into a digital value is associated with an artifact known as read noise. In popular CCD arrays, readout is accomplished in a line-by-line stepwise fashion where pixel values in the first line are read and reset to a pre-determined offset value before shifting the entire array down by a single line in order to read the next line. This procedure is repeated until the entire array has been read out and all the pixels in the chip are reset. Unfortunately, the pixels are never completely cleared, leaving some unknown quantity of residual charge that varies for each pixel and each shifting operation. The result is read (sometimes called reset) noise. Read noise increases with faster readout rates, which becomes a problem with single-molecule imaging experiments requiring fast frame rates. During slow readouts, the read noise can be relatively low (around 2 electrons), whereas at high frame rates of around 20 megapixels per second, the read noise can approach 50 electrons. These high noise values overwhelm the already low (5 to 10 photons) counts at the edge of a single-molecule image, increasing the uncertainty in localization. Therefore, for high-speed readout, large amplification is required as provided by EMCCD cameras (which effectively eliminate read noise). Typically the read noise is the limiting factor for choosing imaging cameras, so the relatively high sensitivity, rapid frame rate, and low noise afforded by EMCCDs are ideal for most PALM applications.
One of the most important considerations for matching a camera to the microscope for single-molecule PALM imaging is to optimize the system magnification. The primary factor that must be addressed is matching the pixel size of the EMCCD camera to the size of the point-spread function. If the pixel size is too large, the point-spread function will be concentrated within too few pixels (undersampled; Figure 10), and there will be insufficient spatial resolution to precisely determine the center. Alternatively, if the pixel size is too small, the point-spread function will be distributed across too many pixels (oversampled) and the signal-to-noise per pixel will be too low for determination of the center. The best choice is a pixel size that is approximately equal to the standard deviation of the point-spread function (approximately 2 pixels; Figure 10, Nyquist Airy disk). The proper magnification can be achieved by use of auxiliary internal tube lenses in microscopes so equipped or through the attachment of an auxiliary magnification system placed between the camera and microscope port. In rough terms, the total magnification for a system using 16-micrometer pixels with a 100x objective with a numerical aperture of 1.49 is approximately 120x (Figure 10), whereas for a 60x objective having the same numerical aperture, the optimum magnification is around 150x.
In summary, the current optimum choice of a suitable detector for single-molecule superresolution using PALM, STORM, or FPALM is an EMCCD that is thermo-electrically cooled to a temperature that minimizes noise artifacts. These advanced camera systems are capable of high frame rates that are not hampered by read noise and are excellent area-array imaging devices to record single molecules. Furthermore, many EMCCD cameras feature a significant flexibility for triggering and exposure when coupled to modern microscope control software packages, enabling the investigator a wide berth of latitude for conducting two-color or live-cell imaging. Perhaps the most important consideration in coupling an EMCCD camera to the microscope is to use the proper intermediate magnification factor so that images of single molecules span a region encompassing a 3 x 3 pixel array. As digital camera technology improves, newer scientific CMOS and high-performance CCDs may eclipse the current lead held by EMCCDs, but that remains to be seen.
back to top ^Computer Hardware and Software Requirements
Because single-molecule superresolution techniques can result in the acquisition of huge datasets, a computer with sufficient random access memory, hard drive storage space, and a fast microprocessor is essential. In addition, the computer must be able to accept hardware interface cards for microscope and camera control. PALM images are typically composed of tens of thousands of single-molecule frames having corresponding file sizes that can easily exceed 5 gigabytes, and therefore require a significant amount of fixed disk storage space. Shuttling data after each acquisition or at the end of the day to a remote server with large storage capacity is recommended whenever possible. Therefore, in planning host computer parameters for a new PALM or STORM system, the investigator should ensure that the computer motherboard is compatible with the selected camera hardware and microscope control requirements (shutters, filter wheels, optical block turret, focus drift correction hardware, etc.). Most of the major manufacturers offer custom software packages or recommended aftermarket products to control microscopes and these should be carefully considered when configuring the instrument. Recording a long series of single-molecule frames can be relatively taxing to the resources on even the fastest computers. Therefore, it is wise to perform localization analysis and image rendering after acquisition using a remote computer equipped with the appropriate analysis software. As PALM software matures with more efficient coding, it should be possible to perform analysis in real-time, thus eliminating the requirement for transporting large datasets to a remote analysis computer.
The basic strategy in rendering PALM images is to plot each molecule as a Gaussian centered at a specific set of coordinates and having a width representing the positional uncertainty or standard error on the measured position of the molecule. In general, the Gaussian width is significantly lower than the positional standard deviation in the original diffraction-limited point-spread function. The analysis of PALM can be described as a series of steps. First, intensity peaks in the raw data corresponding to individual molecules (or fiducial markers) are identified to enable image data corresponding to that molecule to be summed across all frames and pixels in which the molecule appears. Next, the positional coordinates and standard deviation are determined by fitting the summed intensity data to a Gaussian mask according to single-molecule localization theory. If fiducial markers are employed in the experiment, these parameters can be corrected for sample drift during the acquisition (a critical step). Finally, the molecules are rendered together as Gaussians whose brightness indicates the probability that specific molecules can be found at a given location.
Conclusions
When using fluorescent proteins and synthetic fluorophores for single-molecule superresolution experiments, the investigator should monitor for overexpression or excessive staining artifacts while simultaneously attempting to ensure the highest possible molecular density. Localization precision can be affected by the size and type of fluorescent probe used for imaging, with fluorescent proteins and perfectly localized synthetic dyes performing best. Controls should be performed to ensure that the morphological and kinetic characteristics (including shape, growth, and motility) of transfected and stained cells are as close as possible to live specimens imaged in DIC or phase contrast. Transfected and stained cells should be imaged as soon as possible after fixation, as aged specimens tend to slowly lose fluorescence intensity, which leads to a decrease in the final resolution. It is important to note that fixatives (such as paraformaldehyde) should be freshly prepared immediately before use.
During the acquisition period, the most critical parameter to control is the degree of photoactivation at the specimen. In cases where too many molecules are simultaneously photoactivated, it will be difficult to precisely localize a majority of the fluorescent emitters, leading to decreased resolution in the final image. The problem is easy to diagnose by carefully monitoring the acquisition process. If the specimen has an appearance similar to what would be expected under diffraction-limited (widefield) conditions and does not display blinking and photobleaching characteristic of single molecules, the activation power is probably too high and should be reduced until individual emitters appear. Under some circumstances, this problem can be avoided by starting each acquisition with the activation laser turned off until most of the imaged molecules have been bleached. Subsequently, the activation laser can be turned on under very low power. Finally, drift correction is necessary for longer acquisitions to offset the potential for the stage to laterally shift up to 100 nanometers or more during the experiment. Uncorrected drift is manifested in blurry images, but can be corrected using fiduciary markers. Following the suggestions presented above, many of the problems often encountered in PALM imaging can be circumvented or eliminated.
Contributing Authors
Hari Shroff - Section on High Resolution Optical Imaging, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, 20892.
Samuel T. Hess - Department of Physics and Astronomy and Institute for Molecular Biophysics, University of Maine, Orono, Maine, 04469.
Eric Betzig and Harald F. Hess - Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, Virginia, 20147.
George H. Patterson - Biophotonics Section, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda, Maryland, 20892.
Jennifer Lippincott-Schwartz - Cell Biology and Metabolism Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland, 20892.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.